Page 5 of 23
IM10.4-8 | CKD Foundations — SDL Guide (Part 2)
Anaemia of CKD and CKD-Mineral Bone Disorder
Two complications of CKD — anaemia and mineral-bone disease — deserve dedicated study because their pathophysiology is mechanistically distinct, their clinical impact is severe, and their management is a core competency for final-year practice.
Anaemia of CKD is predominantly caused by relative erythropoietin (EPO) deficiency. EPO is produced by peritubular fibroblasts in the renal cortex in response to renal tissue hypoxia; it stimulates erythroid progenitor proliferation in the bone marrow. As nephron mass falls in CKD, EPO production falls disproportionately — a phenomenon attributed to the replacement of EPO-producing fibroblasts by fibrotic tissue. The anaemia is therefore normocytic and normochromic (occasionally mildly hypochromic if concurrent iron deficiency). Additional contributing factors include: (1) shortened red cell survival — uraemic toxins reduce RBC membrane deformability and accelerate haemolysis; (2) functional iron deficiency — hepcidin (an acute-phase protein elevated in CKD due to chronic inflammation) blocks iron absorption from the gut and iron release from macrophages, making stored iron unavailable for erythropoiesis; (3) bone marrow suppression — uraemic toxins and PTH directly inhibit erythroid colony growth; (4) blood losses from frequent phlebotomy, GI uraemic lesions, and dialysis circuits.
The management of anaemia of CKD follows a stepwise approach. First, exclude and correct iron deficiency (check transferrin saturation and ferritin; target ferritin >100 ng/mL and TSAT >20%). Iron should be repleted before starting erythropoiesis-stimulating agents. Then, if Hb remains <10 g/dL despite iron repletion, erythropoiesis-stimulating agents (ESAs) — recombinant human EPO (erythropoietin-alpha, erythropoietin-beta) or darbepoetin alfa — are initiated. The target Hb is 10–11.5 g/dL; targeting Hb ≥13 g/dL with ESAs is associated with increased cardiovascular events (TREAT and CHOIR trial findings). Blood transfusions are avoided in pre-transplant patients (to prevent HLA sensitisation) and are reserved for symptomatic severe anaemia not responsive to ESAs.
CKD-Mineral Bone Disorder (CKD-MBD) is an umbrella term for the interconnected disturbances of mineral metabolism, bone disease, and vascular calcification that develop as GFR falls. The sequence of events begins even in early CKD (G2–G3) and accelerates through G4–G5.
The pathophysiological cascade of CKD-MBD begins with phosphate retention. As the GFR falls, the kidney's capacity to excrete phosphate declines. In the earliest stages, FGF-23 (fibroblast growth factor-23, produced by osteocytes) rises to compensate — it promotes phosphaturia and suppresses 1α-hydroxylase in the proximal tubule (reducing calcitriol production). As GFR falls further, even elevated FGF-23 cannot maintain normal phosphate — hyperphosphataemia develops. Simultaneously, reduced 1α-hydroxylase activity produces calcitriol (1,25-dihydroxyvitamin D) deficiency, which reduces intestinal calcium absorption and directly impairs suppression of PTH secretion. Low calcitriol and low serum calcium (due to reduced intestinal absorption and calcium binding by excess phosphate) drive secondary hyperparathyroidism — PTH rises persistently to mobilise calcium from bone and to promote phosphaturia.
The skeletal consequences of chronically elevated PTH include osteitis fibrosa cystica (bone pain, brown tumours, subperiosteal erosions — characteristic of severe hyperparathyroidism), adynamic bone disease (low-turnover bone disease from oversuppression of PTH with calcitriol/CaCO₃ therapy or in diabetics), and osteomalacia (calcitriol deficiency impairs mineralisation). The vascular consequences are particularly harmful: vascular calcification — driven by hyperphosphataemia and elevated calcium-phosphate product (Ca × PO₄ >55 mg²/dL²) — occurs in the arterial intima (atherosclerotic) and media (Mönckeberg's medial calcification, causing stiff, non-compressible vessels and artificially elevated BP readings). This calcification directly increases cardiovascular mortality.
The management of CKD-MBD targets each step of the cascade: dietary phosphate restriction (limit dairy, nuts, processed foods with phosphate additives); phosphate binders (calcium carbonate or calcium acetate taken with meals — but avoid excess calcium load; non-calcium binders such as sevelamer carbonate are preferred in patients with vascular calcification or hypercalcaemia); calcitriol or vitamin D analogues (alfacalcidol, paricalcitol) to suppress PTH and treat bone disease; and cinacalcet (a calcimimetic that sensitises the calcium-sensing receptor on parathyroid cells — reduces PTH without raising calcium or phosphate) for refractory secondary hyperparathyroidism in dialysis patients.
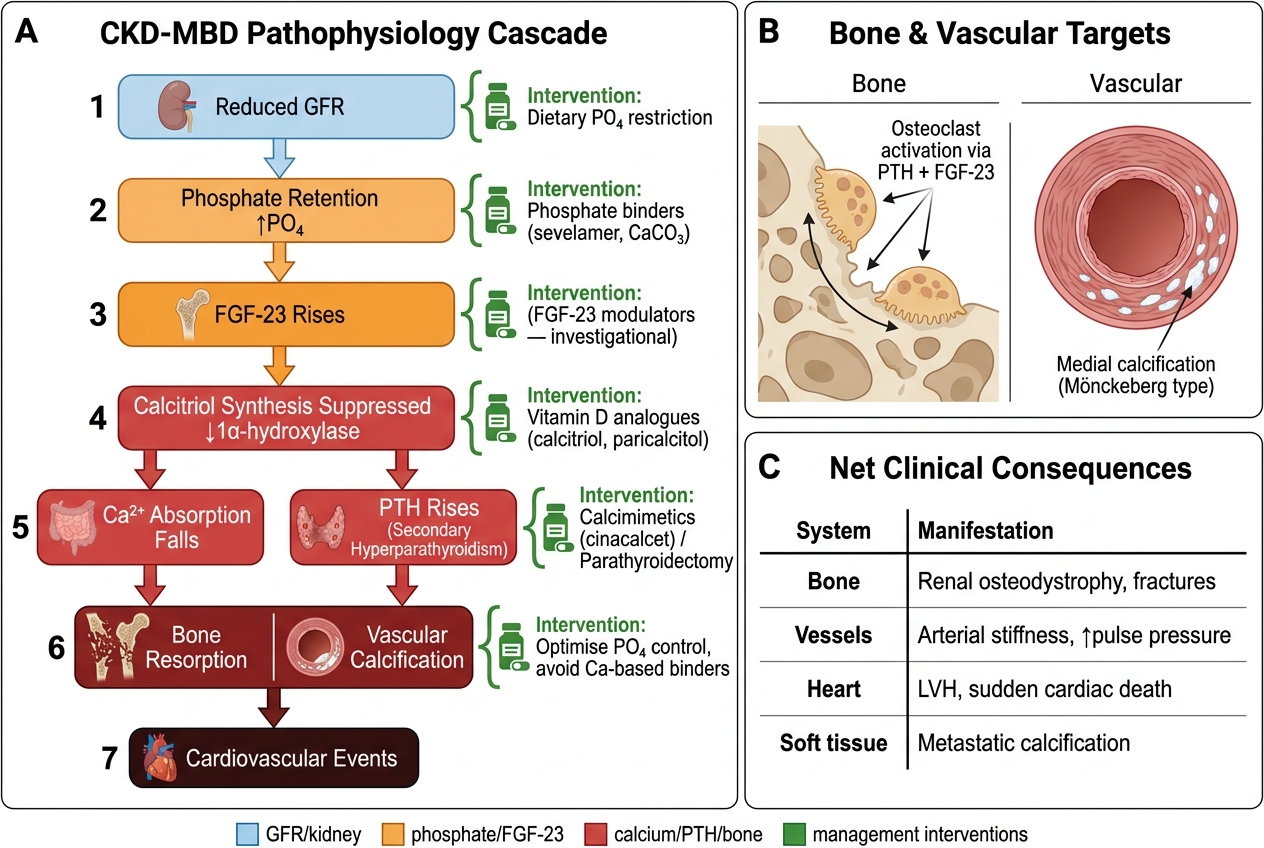
CKD-MBD Pathophysiology Cascade and Management Intervention Points
SELF-CHECK
A patient with CKD G4 has Hb 8.8 g/dL, serum ferritin 45 ng/mL, and transferrin saturation (TSAT) of 15%. His EPO level is disproportionately low for the degree of anaemia. What is the MOST appropriate initial management of his anaemia?
A. Start erythropoiesis-stimulating agent (ESA) immediately
B. Blood transfusion
C. Iron supplementation first, then reassess for ESA
D. High-dose oral folic acid
Reveal Answer
Answer: C. Iron supplementation first, then reassess for ESA
Iron repletion must precede ESA therapy in anaemia of CKD. This patient has functional iron deficiency: ferritin 45 ng/mL (<100 ng/mL target) and TSAT 15% (<20% target) indicate insufficient iron stores for effective erythropoiesis. Starting ESA without iron supplementation will cause an inadequate response and waste an expensive therapy. Once iron is repleted (IV iron preferred in dialysis patients, IV or oral in non-dialysis CKD) and Hb remains <10 g/dL, ESA is initiated. Blood transfusion is avoided in pre-transplant patients. Folic acid is not the primary deficiency.
Diagnosis and Investigation of CKD
The investigation of CKD serves three purposes: confirming the diagnosis and staging, identifying the underlying aetiology (to guide aetiology-specific management and determine reversibility), and screening for complications that accumulate as GFR falls. A structured approach to laboratory and imaging investigations prevents both over-investigation of stable mild CKD and under-investigation of advanced or rapidly progressive disease.
Confirming CKD and staging: The diagnosis requires evidence of kidney damage persisting for >3 months — either a reduced eGFR (<60 mL/min/1.73 m²) or a kidney damage marker such as persistent albuminuria, haematuria of glomerular origin, or structural abnormality. A single creatinine measurement does not establish chronicity; two measurements at least 3 months apart are needed unless there is historical evidence of prior CKD. The CKD-EPI equation using serum creatinine (plus cystatin C where available for greater precision, especially at borderline GFR) generates the eGFR. The spot urine ACR (albumin-to-creatinine ratio, first-morning sample) provides the albuminuria category. Together these two values place the patient in the KDIGO heat map and determine monitoring frequency.
Identifying aetiology: The commonest causes — diabetic nephropathy, hypertensive nephrosclerosis, and chronic glomerulonephritis — can often be inferred clinically, but targeted tests may be needed. A urinalysis with microscopy is essential: glomerulonephritis produces dysmorphic red blood cells and red cell casts; tubular injury produces granular casts; proteinuria can be estimated by dipstick and confirmed by ACR. Renal ultrasound is the first imaging test in all CKD — it assesses kidney size (small, echogenic kidneys confirm chronicity; normal or enlarged kidneys suggest diabetes, amyloid, or polycystic kidney disease), cortical thickness, corticomedullary differentiation, hydronephrosis (obstructive CKD), and cysts. Blood tests to screen for systemic causes include ANA and ANCA (lupus nephritis, vasculitis), serum and urine protein electrophoresis (myeloma), complement levels C3/C4 (membranoproliferative GN, lupus), and hepatitis B and C serology (membranous nephropathy, cryoglobulinaemia). A kidney biopsy is indicated when the cause is unclear, when the presentation is atypical (e.g., rapid GFR decline), or when glomerulonephritis requiring immunosuppression is suspected.
Screening for complications: As GFR falls through the stages, a structured screening panel is repeated at each review visit: serum electrolytes (creatinine, potassium, sodium, bicarbonate); urea (high urea relative to creatinine suggests catabolic state or GI bleeding; the urea:creatinine ratio >20:1 suggests pre-renal contribution); phosphate and calcium (for CKD-MBD monitoring — begin at G3a); PTH and 25-hydroxyvitamin D (secondary hyperparathyroidism); full blood count (anaemia); lipids (cardiovascular risk assessment); and HbA1c in diabetics. The KDIGO monitoring frequency recommendation varies by GFR and albuminuria: G1–G2/A1 = once yearly; G3–G4/A1 = every 6 months; G4–G5 or any stage with A3 = every 3 months or more frequently.
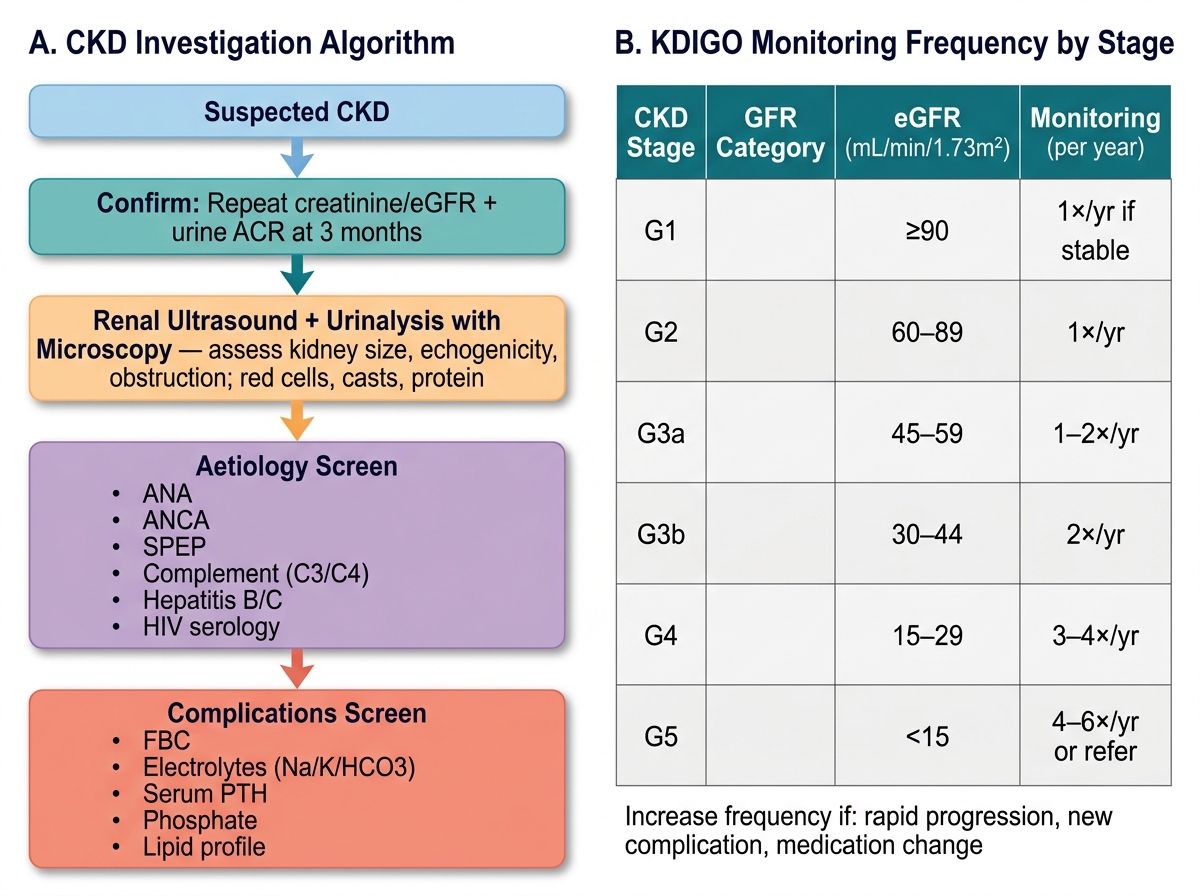
CKD Investigation Algorithm and KDIGO Monitoring Frequency
CKD, Hypertension, and Glycaemia — The Bidirectional Relationship
The triad of CKD, hypertension, and diabetes forms a vicious self-reinforcing cycle in which each element worsens the others. Understanding the bidirectional relationships between these conditions is essential for crafting a coherent management plan that addresses all three simultaneously rather than treating each in isolation.
CKD and Hypertension: Hypertension is both a cause and a consequence of CKD. As a cause, sustained hypertension produces hypertensive nephrosclerosis — afferent arteriolar thickening and glomerular ischaemia leading to glomerulosclerosis and tubular atrophy. In India, hypertensive nephropathy is the second most common cause of ESRD after diabetic nephropathy. As a consequence of CKD, hypertension develops through multiple mechanisms: sodium and water retention (reduced GFR limits sodium excretion, expanding plasma volume), activation of the renin-angiotensin-aldosterone system (RAAS) (ischaemic nephrons release renin, driving angiotensin II-mediated vasoconstriction and aldosterone-mediated sodium retention), and activation of the sympathetic nervous system by uraemic toxins and renal afferent signals. The result is a hypertension that is often sodium-sensitive, volume-driven, and resistant to single-agent therapy. Target blood pressure in CKD is <130/80 mmHg for most patients (regardless of proteinuria presence — 2021 KDIGO guideline update). ACE inhibitors or ARBs are first-line antihypertensives in CKD with proteinuria because they simultaneously lower BP, reduce intraglomerular pressure, and decrease proteinuria. DUAL RAAS blockade (ACE inhibitor + ARB simultaneously) is CONTRAINDICATED — increases risk of hyperkalaemia and AKI without additional renoprotective benefit (ONTARGET trial). In CKD with volume overload, loop diuretics (furosemide) are the diuretic of choice — thiazides lose efficacy when eGFR <30 mL/min/1.73 m².
CKD and Diabetes (Diabetic Nephropathy): Diabetic nephropathy is the commonest cause of ESRD worldwide and the leading single cause in India. The initial lesion is glomerular hyperfiltration (elevated GFR, driven by hyperglycaemia-induced afferent arteriolar dilation), followed by microalbuminuria (A2), then macroalbuminuria (A3), then declining GFR. Histologically, the hallmark findings are Kimmelstiel-Wilson nodules (nodular glomerulosclerosis) and diffuse mesangial expansion. Glycaemic control is the primary strategy for prevention and early retardation of diabetic nephropathy: each 1% reduction in HbA1c reduces microvascular complications (including nephropathy) by approximately 37%. Target HbA1c in CKD is generally <7.0%, but should be relaxed to <8.0% in older patients or those with G4–G5 disease (risk of hypoglycaemia from impaired glucagon response and reduced insulin clearance).
Key pharmacological points for glycaemic management in CKD:
- Metformin: first-line agent for T2DM but must be withheld when eGFR <30 mL/min/1.73 m² (risk of lactic acidosis from impaired renal clearance). Use with caution at eGFR 30–45 (dose-reduce). Stop before iodinated contrast procedures.
- SGLT-2 inhibitors (empagliflozin, dapagliflozin, canagliflozin): now strongly recommended in CKD with or without diabetes — they reduce intraglomerular pressure by promoting natriuresis (relieving tubuloglomerular feedback) and have been shown in landmark trials (CREDENCE, DAPA-CKD, EMPA-KIDNEY) to reduce the risk of ESRD, cardiovascular death, and AKI. Efficacy diminishes at eGFR <25 mL/min (check current indication thresholds per drug), but kidney-protective effects persist to lower GFR values.
- GLP-1 receptor agonists (semaglutide, dulaglutide): also have emerging renal protective data. Dose adjustment needed for some agents in CKD.
- Sulfonylureas: require dose reduction in CKD; risk of prolonged hypoglycaemia (particularly glibenclamide — prefer shorter-acting agents like gliclazide modified release).
- Insulin: doses may need adjustment as CKD progresses (reduced renal insulin clearance can cause hypoglycaemia at previously safe doses).
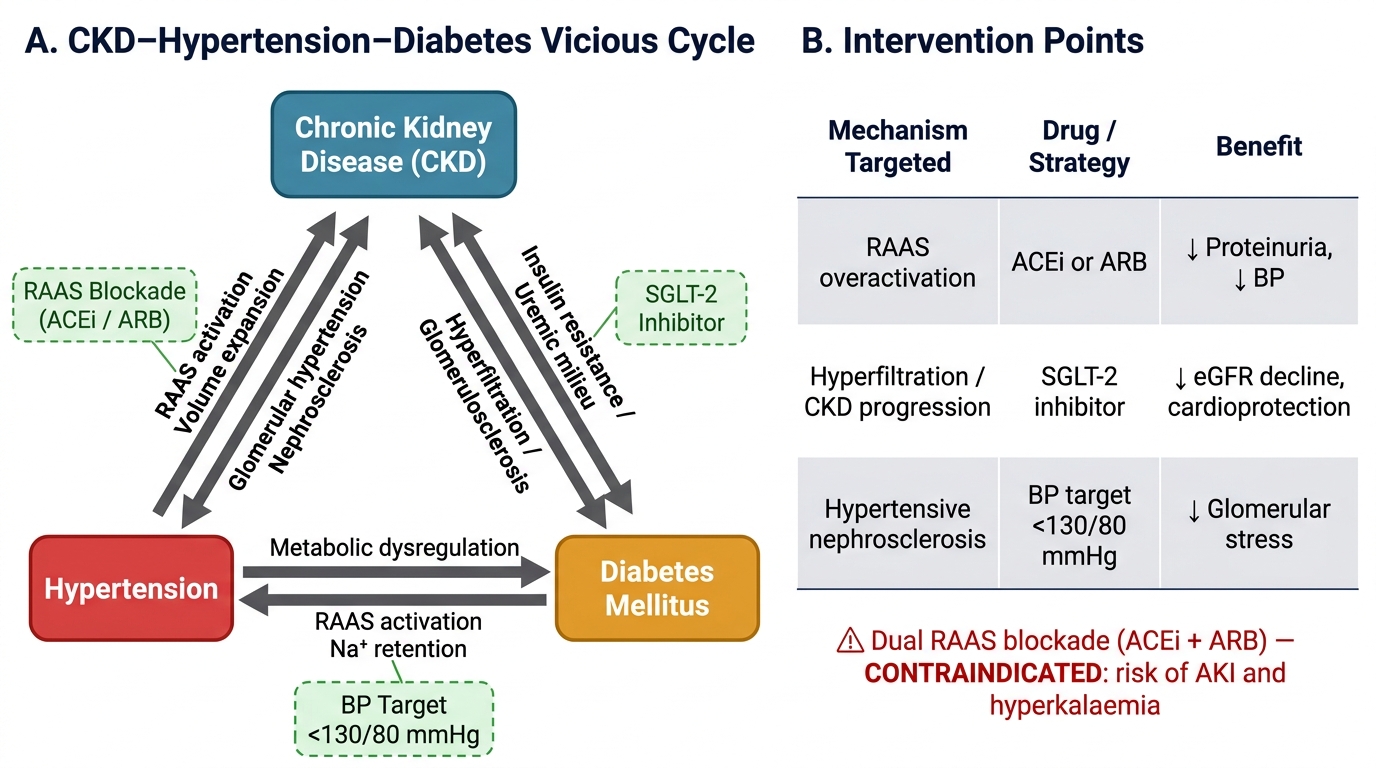
The CKD–Hypertension–Diabetes Vicious Cycle and Key Intervention Points
SELF-CHECK
A 58-year-old type 2 diabetic has eGFR of 42 mL/min/1.73 m², ACR of 220 mg/g, BP 145/88 mmHg, and HbA1c 7.8%. He is currently on metformin 1000 mg twice daily and a calcium channel blocker. Which change to his medication is MOST appropriate?
A. Stop metformin and start an SGLT-2 inhibitor
B. Stop the calcium channel blocker and start dual RAAS blockade with ACE inhibitor plus ARB
C. Add an ACE inhibitor and reduce metformin dose; consider SGLT-2 inhibitor
D. Increase metformin to 2000 mg twice daily for better glycaemic control
Reveal Answer
Answer: C. Add an ACE inhibitor and reduce metformin dose; consider SGLT-2 inhibitor
At eGFR 42 mL/min/1.73 m² (G3b), metformin can be continued but at reduced dose (maximum 500 mg twice daily or as per local guidance — check eGFR regularly). ACE inhibitor or ARB is strongly indicated for CKD with significant albuminuria (ACR 220 mg/g = A2) and hypertension, as they reduce proteinuria and slow CKD progression. SGLT-2 inhibitors (e.g., dapagliflozin, empagliflozin) are now indicated to reduce CKD progression at this eGFR. Dual RAAS blockade (ACE inhibitor + ARB) is contraindicated. Increasing metformin is unsafe. Option C best reflects current evidence-based CKD management combining RAAS blockade, SGLT-2i, and appropriate metformin dose management.
CLINICAL PEARL
The KDIGO CKD heat map is a clinical tool, not just a classification exercise. A patient at G3b/A3 (eGFR 40, ACR 450 mg/g) and a patient at G3b/A1 (eGFR 40, ACR 15 mg/g) have identical GFR categories but profoundly different prognoses — the first has a 40-fold higher risk of progressing to dialysis in 5 years. The A3 patient needs ACE inhibitor/ARB, SGLT-2 inhibitor, more frequent monitoring (every 3 months), and nephrology referral; the A1 patient can be managed in primary care with 6-monthly reviews. Never stage CKD by GFR alone — always add the albuminuria category.
Self-Assessment: Integrating CKD Foundations
You have now covered the complete CKD foundation: the KDIGO two-dimensional staging system combining eGFR categories G1–G5 with albuminuria categories A1–A3; the multisystem pathophysiology and clinical manifestations of uraemia (GI, cardiovascular, neurological, haematological, and skin); the three mechanisms of proteinuria (glomerular, tubular, overflow) and its role as a driver of tubulointerstitial fibrosis and CKD progression; the EPO-deficiency anaemia of CKD and the CKD-MBD cascade from FGF-23 elevation through secondary hyperparathyroidism to vascular calcification; and the bidirectional CKD–hypertension–diabetes cycle with its pharmacological management implications. The self-assessment scenarios below are designed to integrate across these topics and to test your ability to apply the staging system, interpret the laboratory findings, and generate a priority management list — exactly the knowledge needed for the IM10.4–10.8 competencies at the KH level. Read each scenario carefully and formulate your reasoning before examining the analysis.

Systemic Complications of Chronic Kidney Disease (CKD)
Scenario A: A 48-year-old woman with CKD G3b/A3 from IgA nephropathy has BP 138/86 mmHg. Her Hb is 9.8 g/dL, serum ferritin 220 ng/mL, TSAT 28%, and eGFR 35 mL/min/1.73 m². ACR is 480 mg/g. Which two medications are the highest priority?
Analysis: (1) ACE inhibitor or ARB — CKD with A3 albuminuria requires RAAS blockade as the cornerstone nephroprotective treatment. It reduces intraglomerular pressure, decreases proteinuria, and slows CKD progression regardless of BP effect. Target BP <130/80. (2) SGLT-2 inhibitor — CKD G3b is within the indicated range for dapagliflozin (DAPA-CKD: eGFR ≥25 indicated), providing additional eGFR-stabilising benefit. Her anaemia: ferritin 220 and TSAT 28% meet iron sufficiency thresholds — if Hb <10 g/dL persists after 1–3 months of RAAS + SGLT-2, consider ESA.
Scenario B: A 65-year-old man on peritoneal dialysis (ESRD) develops wrist pain and is found to have bilateral subperiosteal erosions of the radial border of the middle phalanges on X-ray. Serum PTH is 890 pg/mL (normal <65), calcium is 10.8 mg/dL, phosphate is 6.2 mg/dL. What is the diagnosis and the most critical immediate management step?
Analysis: Tertiary hyperparathyroidism (or severe secondary hyperparathyroidism refractory to medical management). Subperiosteal erosions of the phalanges are pathognomonic of osteitis fibrosa cystica. The calcium is elevated (hypercalcaemia from autonomous PTH secretion) and phosphate is very high. Immediate: (1) Use non-calcium phosphate binders (sevelamer) — calcium-based binders would worsen hypercalcaemia; (2) Stop calcitriol (further raising calcium); (3) Add cinacalcet — reduces PTH by sensitising the calcium-sensing receptor; (4) If refractory to cinacalcet over 6 months, refer for parathyroidectomy.
Scenario C: A 55-year-old with CKD G4 and type 2 diabetes reports increasing fatigue and tingling in both feet. His eGFR is 23 mL/min/1.73 m², Hb 9.0 g/dL. What uraemic complications does this presentation suggest, and what screening test would confirm the neurological finding?
Analysis: Two uraemic complications: (1) Anaemia of CKD — EPO deficiency at eGFR 23; (2) Uraemic peripheral neuropathy — stocking-pattern tingling in both feet at GFR 23 mL/min. Screening test: nerve conduction velocity studies (NCV) showing sensorimotor neuropathy with reduced conduction velocity confirms uraemic neuropathy (important to distinguish from diabetic peripheral neuropathy — the two can coexist and NCV pattern is similar; clinical context and response to dialysis differentiation matter).