Page 2 of 14
IM19.1-2 | Movement Disorder Foundations — SDL Guide (Part 2)
Diagnostic Framework: Applying the Classification to Clinical Presentations
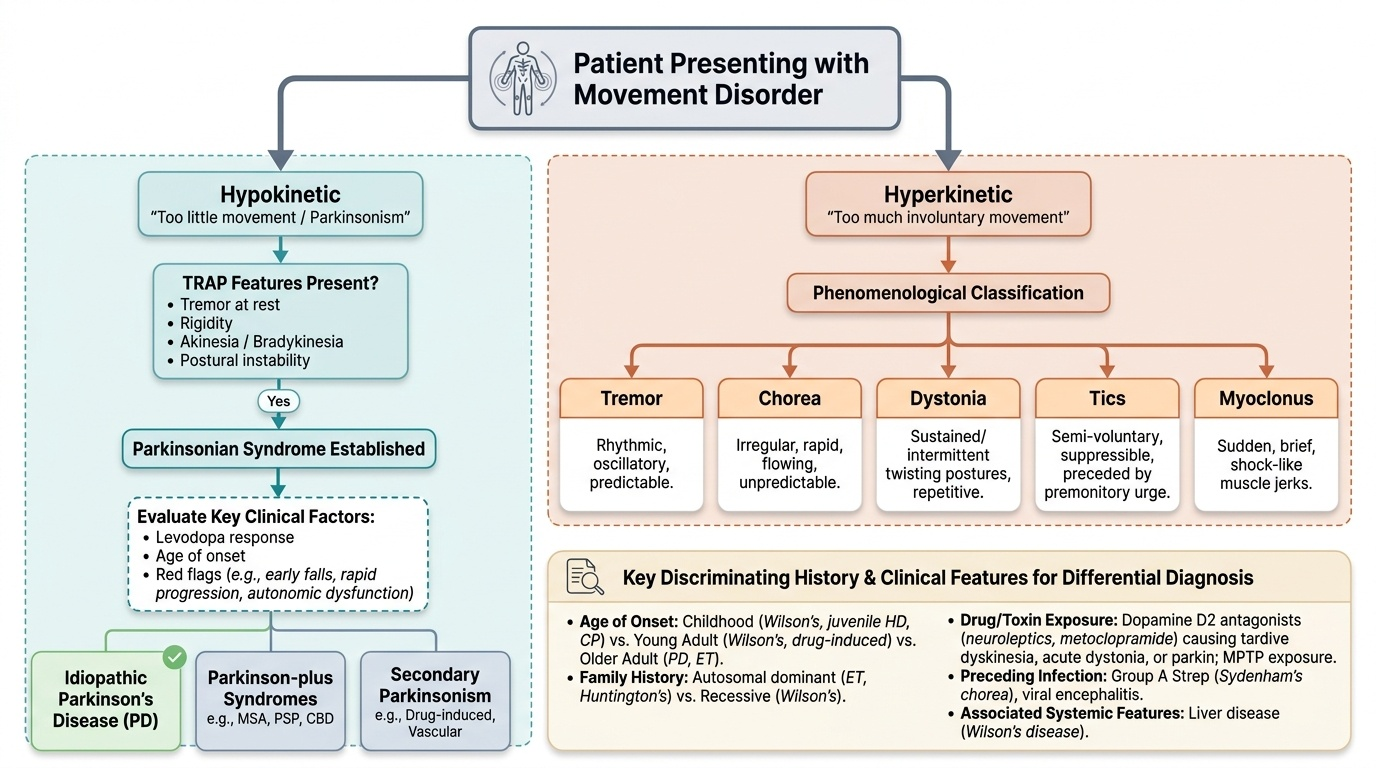
The classification criteria described above — distribution, rhythm, repetition, activation, and exacerbating/relieving factors — are not merely academic. They directly determine the initial diagnostic evaluation and the differential diagnosis. A systematic approach to characterising the movement disorder phenomenologically precedes all investigations and narrows the differential far more efficiently than an undirected workup.
Provided image
The first clinical task is to establish the predominant phenomenology: is the primary problem hypokinesia (too little movement — parkinsonism) or hyperkinesia (too much involuntary movement)? This distinction orients the entire clinical approach. In the hypokinetic category, the presence of the TRAP features (tremor at rest, rigidity, akinesia/bradykinesia, postural instability) establishes a parkinsonian syndrome; the subsequent question is the cause (idiopathic PD vs secondary vs Parkinson-plus). In the hyperkinetic category, the character of the involuntary movement — its rhythmicity, its predictability, its relationship to voluntary action, and whether it can be suppressed — determines the category (chorea, dystonia, tics, myoclonus, tremor).
Key discriminating questions for history:
- Age of onset: childhood/adolescent onset → Wilson's disease, Huntington's (juvenile), cerebral palsy, primary dystonia; young adult → Wilson's, drug-induced, Huntington's early; older adult → PD, essential tremor, drug-induced.
- Family history: essential tremor (autosomal dominant), Huntington's (dominant), Wilson's disease (autosomal recessive), DYT1 dystonia (dominant).
- Drug and toxin exposure: neuroleptics (dopamine D2 antagonists) — first-generation antipsychotics (haloperidol, chlorpromazine), metoclopramide, domperidone — cause drug-induced parkinsonism, tardive dyskinesia (repetitive oral-facial movements, often with trunk and limb involvement — develops after months to years of exposure), acute dystonic reaction (within hours-days), and akathisia (restlessness). MPTP (a meperidine analogue contaminant used by drug abusers) destroys SNc neurons and causes rapid-onset parkinsonism.
- Preceding infection: sore throat + chorea in a child → Sydenham's chorea; encephalitis → movement disorder.
- Associated systemic features: liver disease + movement disorder + psychiatric symptoms in a young person → Wilson's disease (see below).
Wilson's disease (hepatolenticular degeneration) is a crucial diagnostic trap in young patients with movement disorders. It is an autosomal recessive disorder of copper transport (ATP7B gene mutation), resulting in copper accumulation in liver, brain (basal ganglia and thalamus), cornea, and kidneys. Any young patient (<40 years) presenting with a movement disorder AND liver disease should be investigated for Wilson's. The movement disorder can be varied: tremor (wing-beating tremor on arms-outstretched is classic), dysarthria, dystonia, parkinsonism, or choreoathetosis. The hallmark examination finding is the Kayser-Fleischer (KF) ring — a golden-brown ring at the corneal periphery (Descemet's membrane) visible on slit-lamp examination; present in virtually all neurological Wilson's. Laboratory confirmation: low serum ceruloplasmin (<20 mg/dL; the normal is 20–40), elevated 24-hour urinary copper (>100 μg/24h in symptomatic patients; >250 μg/24h diagnostic), and liver biopsy showing elevated hepatic copper. Genetic testing for ATP7B mutations confirms the diagnosis.
SELF-CHECK
A 22-year-old man presents with a 10-month history of tremor, dysarthria, and personality change. He has jaundice and hepatomegaly on examination. Which combination of investigations would BEST confirm the diagnosis?
A. MRI brain and serum copper alone
B. Serum ceruloplasmin, 24-hour urinary copper, and slit-lamp examination for KF rings
C. DAT-SPECT scan and serum dopamine
D. EEG and antinuclear antibody titre
Reveal Answer
Answer: B. Serum ceruloplasmin, 24-hour urinary copper, and slit-lamp examination for KF rings
The triad of movement disorder + psychiatric change + liver disease in a young patient is Wilson's disease until proved otherwise. The diagnostic combination is: low serum ceruloplasmin (<20 mg/dL), elevated 24-hour urinary copper (>100 μg/24h in symptomatic patients, >250 μg/24h diagnostic), and Kayser-Fleischer (KF) rings on slit-lamp examination (virtually universal in neurological Wilson's). MRI brain may show T2 hyperintensity in the basal ganglia ('face of the giant panda' sign on axial T2 through the midbrain — a recognised pattern) but is not the primary confirmatory test. Serum copper can be normal or even elevated and is not a reliable standalone test.
Management Principles: How Classification Steers Treatment
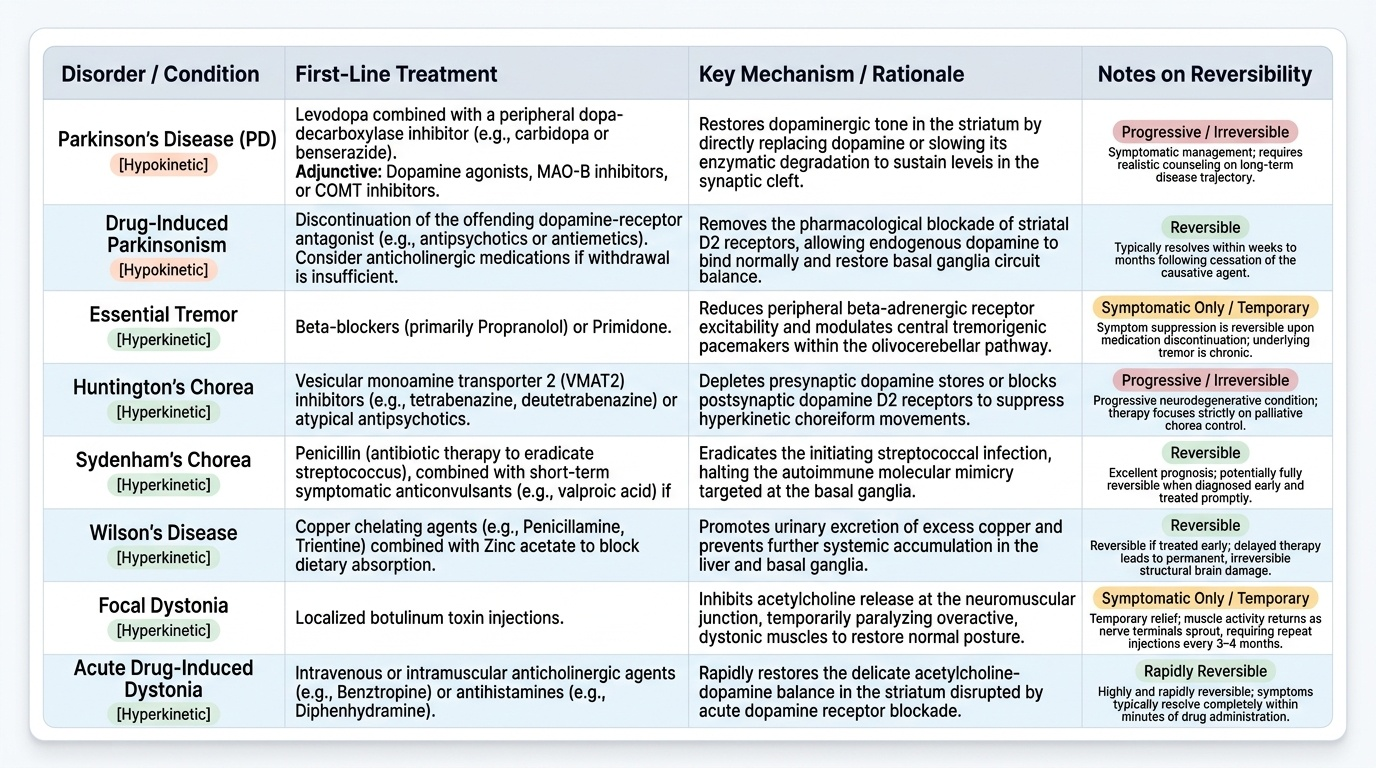
Management of movement disorders is tightly coupled to their classification — the phenomenological category, the underlying aetiology, and the anatomical substrate all determine the therapeutic approach. There is no single treatment for 'movement disorder'; rather, the classification framework developed in the preceding sections directly steers the treatment decision. This section provides an overview of therapeutic principles for each major category; detailed pharmacology of specific agents (especially for Parkinson's disease) is covered in the SDL on Movement Disorder Investigation and Parkinson Therapy.
Provided image
The fundamental principle underlying all treatment selection is this: once you have identified whether the disorder is hypokinetic or hyperkinetic, and determined its specific aetiology, the therapeutic target becomes clear. For hypokinetic disorders, treatment generally aims to restore dopaminergic tone in the striatum — either by replacing dopamine (levodopa), stimulating dopamine receptors (agonists), or slowing its degradation (MAO-B inhibitors, COMT inhibitors). For hyperkinetic disorders, the approach depends on the aetiology: remove the offending cause where identifiable (a drug, a copper metabolic defect, a streptococcal infection), suppress the abnormal movement with pharmacological agents that reduce dopaminergic or thalamo-cortical excitability, or modulate the circuit surgically when pharmacotherapy is insufficient. The reversibility of the underlying process is a critical consideration — Wilson's disease and Sydenham's chorea are potentially reversible if treated promptly, while Huntington's disease and idiopathic PD are progressive and require symptomatic management alongside realistic patient counselling about long-term trajectory.
Hypokinetic disorders — parkinsonian syndromes:
- Idiopathic Parkinson's disease: the foundation of pharmacological management is dopamine replacement or dopamine receptor stimulation. Levodopa (combined with a peripheral dopa-decarboxylase inhibitor — carbidopa or benserazide — to reduce peripheral conversion and side effects) remains the most effective symptomatic treatment. Dopamine agonists (pramipexole, ropinirole, rotigotine), MAO-B inhibitors (selegiline, rasagiline), and COMT inhibitors (entacapone, tolcapone) are adjunctive agents. Non-pharmacological management includes physiotherapy, occupational therapy, speech therapy, and, in advanced disease, deep brain stimulation (DBS) of the STN or GPi.
- Secondary parkinsonism: remove the offending cause where possible (stop the offending drug in drug-induced parkinsonism; treat Wilson's disease with chelation — D-penicillamine or trientine — or zinc supplementation; manage vascular risk factors in vascular parkinsonism). Levodopa may help in some secondary forms but is less effective than in idiopathic PD.
- Parkinson-plus syndromes (PSP, MSA, CBS): levodopa response is typically poor; management is largely symptomatic (physiotherapy, falls prevention, dysphagia management, autonomic support for MSA).
Hyperkinetic disorders:
- Essential tremor: first-line pharmacotherapy is propranolol (beta-blocker — suppresses enhanced physiological component, 50–70% response) or primidone (anticonvulsant, effective at low doses for tremor). DBS of the ventral intermediate nucleus (VIM) of the thalamus is effective for medically refractory essential tremor.
- Chorea — Huntington's disease: there is no disease-modifying treatment currently available. Chorea can be suppressed symptomatically with tetrabenazine (vesicular monoamine transporter-2 [VMAT2] inhibitor — depletes presynaptic dopamine) or deutetrabenazine; neuroleptics (haloperidol, clonazepam) are alternatives. Genetic counselling and psychological support are essential.
- Sydenham's chorea: the chorea is self-limited (resolves in weeks to months with anti-streptococcal treatment and penicillin prophylaxis for ARF prevention). If chorea is disabling, valproate or carbamazepine can be used to suppress it.
- Wilson's disease: copper chelation — D-penicillamine (first-line historically; associated with initial neurological deterioration in ~20%) or trientine (preferred if neurological disease, fewer side effects); zinc acetate/sulphate blocks intestinal copper absorption (used for maintenance and in pre-symptomatic patients). Dietary restriction of copper-rich foods (liver, shellfish, nuts, chocolate). Liver transplantation corrects the hepatic copper metabolism defect and may stabilise or improve neurological disease.
- Drug-induced movement disorders: immediately cease or reduce the offending dopamine-blocking agent if possible. Acute dystonic reaction → anticholinergics (procyclidine, benztropine). Tardive dyskinesia → VMAT2 inhibitors (tetrabenazine, deutetrabenazine, valbenazine); time-limited improvement often occurs after drug cessation but tardive dyskinesia can be irreversible.
- Focal dystonias: botulinum toxin injection into the affected muscle group is the first-line treatment for focal dystonia (cervical dystonia, blepharospasm, writer's cramp); provides 3–4 months of benefit per injection cycle. Generalised dystonia: oral anticholinergics (trihexyphenidyl), baclofen (especially intrathecal for severe cases), or DBS (GPi is the target for dystonia).
SELF-CHECK
A 35-year-old woman with schizophrenia has been on haloperidol for 4 years. She develops repetitive, involuntary lip-smacking, tongue-rolling, and facial grimacing that her psychiatrist notices at a routine review. What is this movement disorder and what is the first management step?
A. Acute dystonic reaction — administer intramuscular procyclidine immediately
B. Neuroleptic malignant syndrome — stop haloperidol and admit to ICU
C. Tardive dyskinesia — reduce or stop the offending neuroleptic where clinically safe
D. Essential tremor — add propranolol to the existing regimen
Reveal Answer
Answer: C. Tardive dyskinesia — reduce or stop the offending neuroleptic where clinically safe
Tardive dyskinesia (TD) is a hyperkinetic movement disorder characterised by repetitive, involuntary orofacial movements (lip-smacking, tongue protrusion, facial grimacing) — and can also involve the trunk and limbs — developing after months to years of dopamine receptor-blocking agent (DRBA) exposure. It is distinct from acute dystonic reactions (which occur within hours to days, present with sustained muscle contraction/posturing, and respond to anticholinergics) and from akathisia (subjective restlessness with repetitive purposeless movements). The first management step is to reduce or (if clinically safe) stop the offending DRBA. VMAT2 inhibitors (tetrabenazine, valbenazine, deutetrabenazine) can suppress TD; some TD is irreversible even after drug cessation.
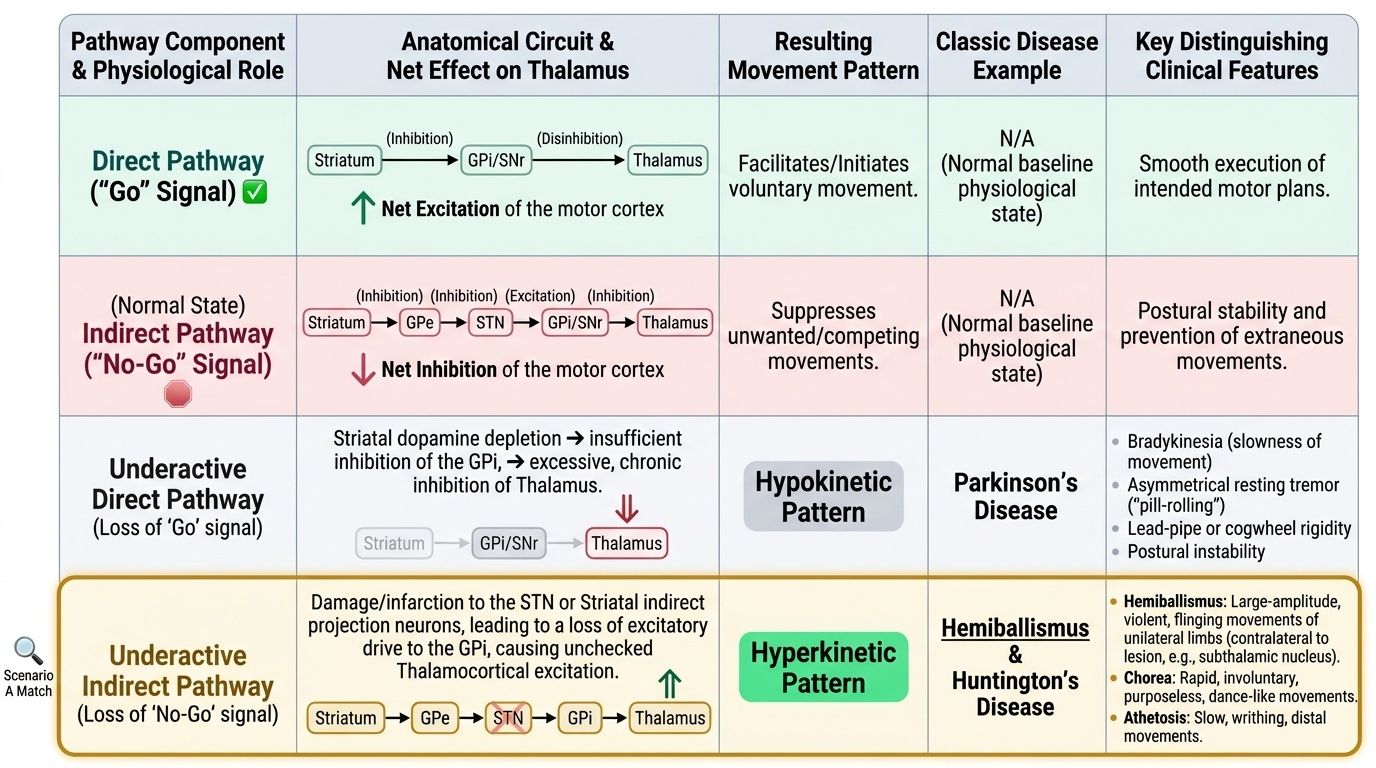
Self-Assessment: Integrating Neuroanatomy and Classification
At this stage in the module you have built a coherent model of how the basal ganglia circuit works, how its disruption at specific nodes produces hypokinetic or hyperkinetic disorders, and how the phenomenological classification — using distribution, rhythm, repetition, activation, and modifying factors — brings order to a clinically diverse group of conditions. The self-assessment scenarios below challenge you to apply the circuit model to a clinical presentation and arrive at the correct phenomenological category and mechanism before reading the analysis. This skill — moving from clinical observation to anatomical localisation — underpins the NMC competency IM19.1 and IM19.2 at the KH level. For each scenario, read the clinical context carefully, identify the key discriminating feature in the history or examination, localise the anatomical substrate using the circuit model, and reach the diagnosis before reading the analysis. This is the transfer of learning from declarative knowledge (knowing the circuit) to procedural reasoning (applying it to a patient) — the core competency tested in IM19.1 and IM19.2 at the KH level required by the NMC curriculum.
Provided image
Scenario A: A 45-year-old man develops large-amplitude, violent, flinging movements of his right arm after a small stroke. On CT head, there is a hypodense infarct in the left basal ganglia region. What anatomical structure is most likely damaged and through which circuit mechanism does the movement arise?
Analysis: The movement is hemiballismus — violent, large-amplitude, proximal limb movements on one side (contralateral to the lesion). The responsible lesion is in the contralateral subthalamic nucleus (STN). Loss of STN activity reduces its excitatory drive on GPi → GPi becomes underactive → thalamus is disinhibited → excessive, uncontrolled thalamo-cortical motor discharge → hemiballismus. This is a direct clinical illustration of the indirect pathway: STN normally excites GPi which inhibits thalamus; destroy the STN and the thalamus is released.
Scenario B: A 17-year-old girl develops sudden-onset, random, dance-like movements of her limbs and emotional lability two weeks after a bout of tonsillitis. Throat swab culture is negative, but ASOT titre is elevated. Her ECG shows prolonged PR interval. What is the diagnosis, what is the mechanism, and what other organ must be examined carefully?
Analysis: Sydenham's chorea — post-streptococcal autoimmune movement disorder associated with acute rheumatic fever. The mechanism is molecular mimicry: anti-streptococcal antibodies cross-react with basal ganglia neurons (particularly the caudate and putamen). The ECG finding (prolonged PR = first-degree AV block) is part of acute rheumatic fever (Duckett Jones criteria). The heart must be examined carefully for carditis (new murmur → valvular disease, most commonly mitral regurgitation). Management: penicillin/amoxicillin to eradicate streptococcus; secondary prophylaxis with long-acting benzathine penicillin monthly; treat cardiac involvement; chorea usually resolves spontaneously.
Scenario C: A 28-year-old man presents with bilateral arm tremor present only when he holds his arms outstretched against gravity. The tremor is regular, 8 Hz, fine, and bilateral. His father had the same tremor. His neurological examination is otherwise entirely normal. What is the most likely diagnosis, and which medication is first-line?
Analysis: Essential tremor — the commonest movement disorder in adults; postural tremor (present with sustained posture, absent at rest, not worsening on target approach), bilateral, with a positive family history (autosomal dominant). The tremor frequency is typically 6–12 Hz. Distinguished from PD (PD tremor is rest tremor, asymmetric, 4–6 Hz, with bradykinesia and rigidity). First-line pharmacotherapy: propranolol 40–320 mg/day (beta-blocker) or primidone 50–750 mg/day (anticonvulsant). Alcohol transiently suppresses essential tremor (not a treatment but a clue in the history).
CLINICAL PEARL
The single most important clinical distinction in movement disorders is the activation condition of tremor. Rest tremor (disappears with voluntary movement) = basal ganglia disease → Parkinson's disease until proved otherwise. Intention tremor (worsens on target approach) = cerebellar disease. Postural tremor (present with sustained posture, absent at rest) = essential tremor, physiological, or drug-induced. Many students erroneously assume that any tremor is Parkinson's disease — but treating essential tremor with levodopa is ineffective, and missing a cerebellar cause has entirely different prognostic and management implications. Ask one question: 'Does the tremor get worse when you reach for something, or better?'
A second pearl specific to India: in any patient under 40 with a movement disorder, always screen actively for Wilson's disease. It is treatable and reversible if caught early, but rapidly progressive and potentially fatal if missed. The clinical triad of movement disorder + psychiatric symptoms + liver disease in a young person should trigger slit-lamp examination and ceruloplasmin/24-hour urinary copper without delay.