Page 11 of 24
IM2.{14-16,19-20,23} | ACS Acute Management — SDL Guide
Learning Objectives
- Describe the indications for CCU admission and the components of supportive care in ACS
- Discuss the medications used in ACS — antiplatelet agents, anticoagulants, nitrates, beta-blockers, ACE inhibitors, and GPIIb/IIIa inhibitors — with their indications, contraindications, and mechanisms
- Describe the indications for thrombolysis, primary PCI, and CABG in ACS
- Describe the pathogenesis, recognition, and management of ACS complications — arrhythmias, cardiogenic shock, LV dysfunction, papillary muscle rupture, and pericarditis
- Discuss the assessment and relief of pain in ACS
INSTRUCTIONS
This module covers the full arc of ACS acute management: from the moment of admission through CCU supportive care, pharmacotherapy, reperfusion decision-making, complication management, and pain control. Every drug and procedural decision has a specific indication, contraindication, and timing window that must be understood mechanistically, not merely memorised.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 295 — Ischaemic Heart Disease (textbook)
- API Textbook of Medicine, 10th ed., Ch. on ACS Management (textbook)
- ESC 2023 Guideline for Acute Coronary Syndromes (guideline)
- ACC/AHA 2022 Guideline for Chest Pain and ACS Management (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Suresh, the 52-year-old diabetic smoker from the first module in this cluster, arrives in the ED at 9 PM with 2 hours of crushing chest pain. His ECG shows 4-mm ST elevation in V1–V4 with reciprocal depression in II, III, and aVF. His BP is 100/70 mmHg, HR 110/min, SpO2 93%. This is an anterior STEMI. The decision tree for the next 90 minutes will determine whether Suresh survives with preserved LV function or dies from cardiogenic shock. Every decision — aspirin or no aspirin, which antiplatelet to load, thrombolysis or PCI, morphine dose, whether to start heparin now — has a correct answer grounded in evidence. Suresh's BP of 100/70 mmHg is a particular challenge: low enough to raise concern about cardiogenic shock, but also a contraindication to certain drugs. How do you navigate that? And if, on day 3, the nurse reports a new harsh pansystolic murmur and Suresh's SpO2 has fallen to 86% — what mechanical complication has occurred, what is its mechanism, and how urgently does he need surgery? This module equips you with the clinical decision-making framework for every management step in acute ACS.
WHY THIS MATTERS
The competency set IM2.14–2.23 requires knowledge-and-higher (KH) level understanding of ACS management — meaning you must not only recall the drugs and doses but be able to apply them to patient scenarios, identify contraindications, recognise complications, and justify management decisions. For the final-year student in India, this is particularly consequential: you will rotate through medicine wards, ICUs, and emergency departments where ACS patients are actively managed. The ability to initiate evidence-based supportive care — aspirin loading, anticoagulation, morphine titration, nitrate administration, CCU triage — while you wait for the cardiologist is not optional. It is the expected standard of a provisionally registered medical practitioner.
RECALL
Recall from preceding modules: STEMI is treated as an emergency requiring reperfusion (primary PCI or thrombolysis); NSTEMI/UA requires risk stratification and an early invasive strategy (coronary angiography within 24–72 hours). The Killip classification (I–IV) defines haemodynamic severity — Killip IV (cardiogenic shock) demands the most intensive intervention. From pharmacology, recall: aspirin inhibits COX-1, blocking thromboxane A2-mediated platelet aggregation irreversibly; heparin potentiates antithrombin III to inhibit factor Xa and thrombin; morphine acts on μ-opioid receptors to provide analgesia and reduce preload (venodilation). Keep these mechanisms in mind as you build the evidence-based rationale for each drug class in ACS.
Clinical Presentation of ACS Complications
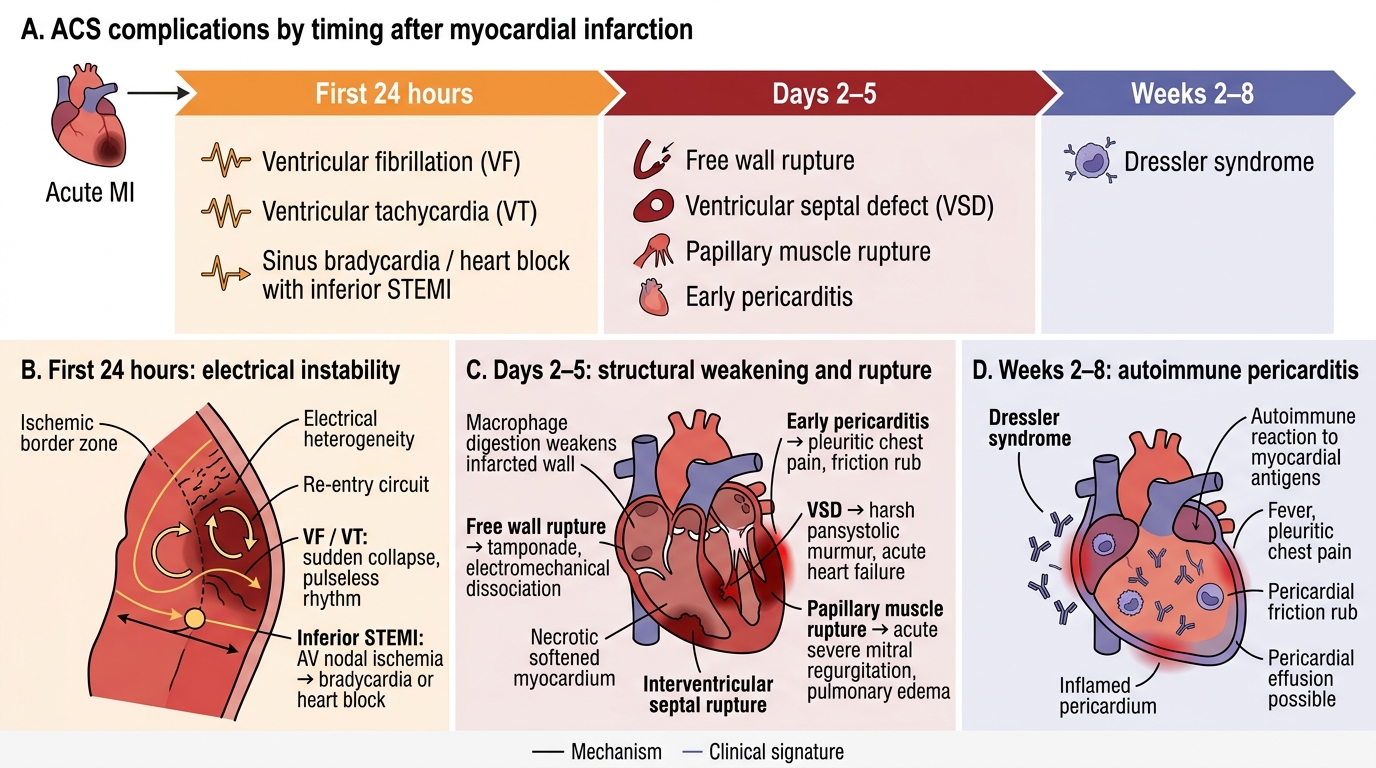
Understanding the clinical presentations of ACS complications is the foundation for recognising them at the bedside — before haemodynamic collapse makes intervention more difficult. The complications of ACS can be classified into electrical (arrhythmias and conduction disturbances), mechanical (structural failures of the heart), and inflammatory/pericardial (pericarditis). Each has a characteristic timing after MI onset, a pathological mechanism directly tied to the infarct territory, and a clinical presentation that is recognisable if you know what to look for. The electrical complications tend to dominate the first 24 hours, the mechanical complications emerge on days 2–5 as the necrotic myocardium undergoes enzymatic softening, and the pericardial complications span from day 1 (early epistenocardial pericarditis) to weeks 2–8 (Dressler syndrome). Knowing this temporal pattern converts complication recognition from memorisation into pattern-based clinical reasoning — if it is day 3 and a new murmur appears, you are immediately thinking papillary muscle rupture or VSD; if it is day 1 and the patient has pleuritic pain, you are thinking early pericarditis.
Electrical complications — arrhythmias:
- Ventricular fibrillation (VF): the most common cause of sudden death in the first 24 hours of STEMI, due to re-entry circuits in the border zone of ischaemic and non-ischaemic myocardium. Presents as sudden haemodynamic collapse without a palpable pulse; ECG shows chaotic high-amplitude waveforms with no discernible P or QRS complexes. Immediate defibrillation is the only effective treatment — precordial thump is no longer recommended (Class III in current guidelines). CPR while charging the defibrillator.
- Ventricular tachycardia (VT): monomorphic VT (regular wide-complex tachycardia at >100/min) in the context of acute MI indicates significant myocardial ischaemia. Sustained VT >30 seconds or causing haemodynamic compromise requires synchronised DC cardioversion. Lidocaine (1–1.5 mg/kg IV bolus) or amiodarone (150 mg IV over 10 minutes) are pharmacological options for termination or suppression.
- Accelerated idioventricular rhythm (AIVR): a regular wide-complex rhythm at 60–100/min occurring in the first 12–24 hours post-MI or after successful reperfusion — the 'reperfusion arrhythmia'; usually benign and self-terminating; does NOT require treatment and should not be confused with VT (rate distinguishes them)
- Sinus bradycardia with inferior STEMI: Bezold-Jarisch reflex (vagal activation) causes sinus bradycardia or Mobitz type I (Wenckebach) AV block — usually resolves with reperfusion; atropine (0.5 mg IV, repeated to 3 mg total) if symptomatic bradycardia (HR <40, haemodynamic compromise). Complete heart block (CHB) with inferior STEMI: indicates involvement of the AV nodal artery (branch of RCA); usually transient with reperfusion but may require temporary pacing (transcutaneous pacing as bridge, then transvenous if persistent).
- New LBBB or complete heart block with anterior STEMI (LAD occlusion with septal involvement): indicates bilateral bundle branch block territory infarction; high risk of progression to complete heart block or asystole; prophylactic temporary pacemaker required.
- Atrial fibrillation (new onset): occurs in ~10% of STEMI patients, usually in the context of LV dysfunction or haemodynamic compromise; associated with increased thromboembolic risk (anticoagulation) and rate or rhythm control as indicated
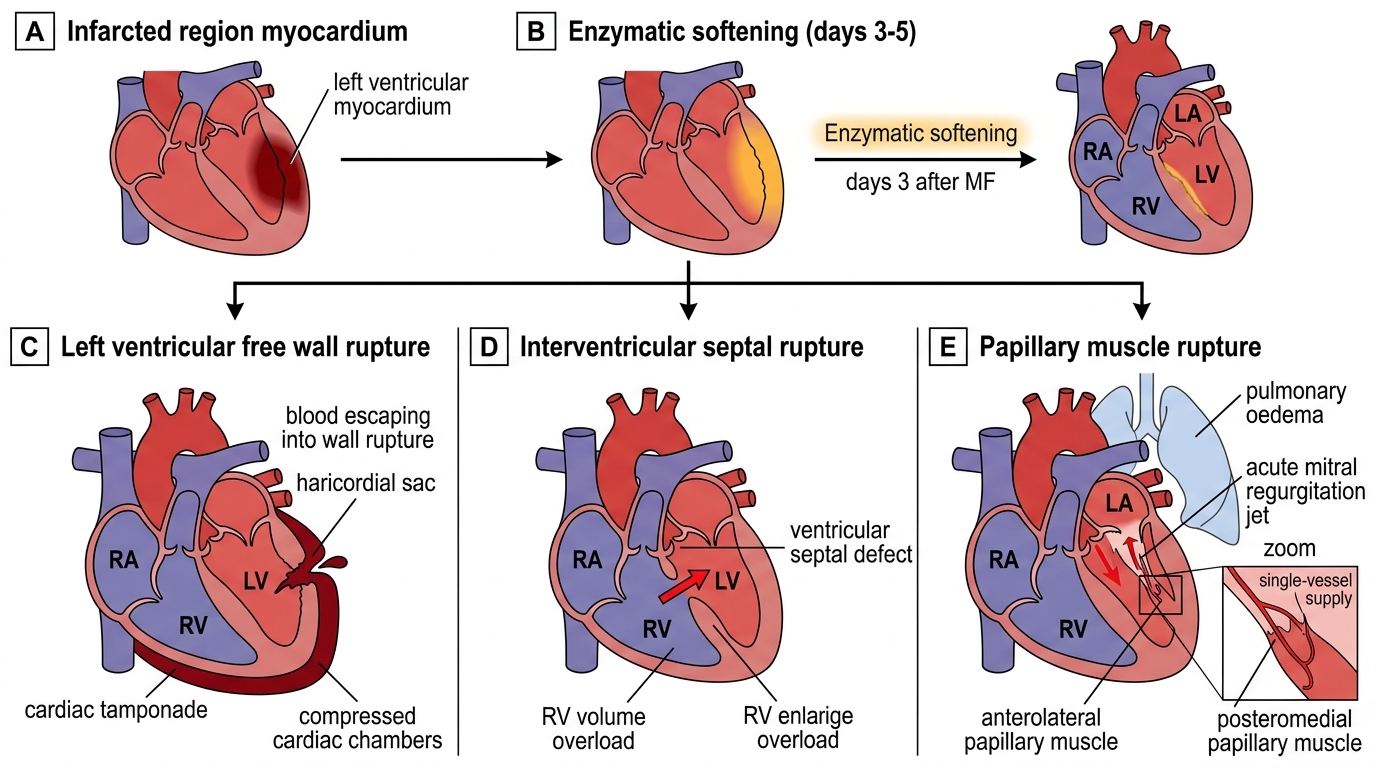
Mechanical complications — these are life-threatening structural failures occurring 2–5 days post-MI (peak at day 3–5 as necrotic myocardium undergoes autolytic softening):
- Free wall rupture: catastrophic — sudden haemodynamic collapse, electromechanical dissociation (EMD/PEA on monitor, organised QRS without pulse), cardiac tamponade (elevated JVP, hypotension, muffled heart sounds — Beck's triad). Rare (1–3% of untreated STEMI), more common in older women, first MI, anterior location, and hypertension. Emergency pericardiocentesis for temporising, followed by immediate surgical repair.
- Ventricular septal defect (VSD): septal rupture due to necrosis of the interventricular septum (proximal LAD territory). Presents as sudden haemodynamic deterioration with a new harsh pansystolic murmur at the left sternal border, confirmed by echo (colour Doppler shows L→R shunt across the septum). Surgical repair or transcatheter closure urgently after initial stabilisation with vasodilators (if not in shock) and intra-aortic balloon pump.
- Papillary muscle rupture: ischaemia of the posteromedial papillary muscle (single RCA supply, unlike anterolateral which has dual supply) causes partial or complete rupture, producing acute mitral regurgitation. Presents as a new pansystolic murmur at the apex radiating to the axilla, with acute pulmonary oedema (the regurgitation is acute, the LA has no time to dilate, LV pressure transmitted backwards). Surgical mitral valve repair/replacement urgently. Echo differentiates from VSD by murmur quality and colour Doppler findings.
Pericardial complications:
- Early pericarditis (epistenocardial pericarditis): inflammatory reaction of the pericardium overlying the infarcted zone, occurring on day 1–2. Presents as sharp, pleuritic chest pain (worse supine, better sitting forward), pericardial friction rub on auscultation. ECG: widespread saddle-shaped ST elevation (but this may be difficult to distinguish from residual STEMI changes in the acute phase). Treated with high-dose aspirin (1000 mg TDS × 2 weeks) or colchicine (0.5 mg BD × 3 months) — NSAIDs other than aspirin are avoided in the acute MI phase as they impair myocardial healing.
- Dressler syndrome: autoimmune pericarditis occurring 2–8 weeks post-MI (immune complex deposition in the pericardium). Presents with fever, pericarditic chest pain, pleural effusion, pericardial rub. Treat with NSAIDs or colchicine; corticosteroids if refractory. Anticoagulation risk increases with large effusion.
Timeline of ACS Complications After MI
Pathophysiology: Why Each ACS Complication Occurs
Each ACS complication has a specific pathophysiological mechanism tied to the territory and depth of infarction, the time course of myocardial necrosis, and the structure-specific vulnerability of different cardiac components. Understanding these mechanisms is not merely academic — it predicts which complication is most likely in a given clinical scenario and explains why the management of each complication differs fundamentally.
Arrhythmic mechanism: In the first minutes to hours of coronary occlusion, the ischaemic zone generates electrical heterogeneity — adjacent cells have different resting potentials, action potential durations, and refractory periods. This heterogeneity creates re-entry circuits in the peri-infarct border zone, which are the substrate for VF and VT. The longer the ischaemia, the more extensive the zone of heterogeneity and the greater the arrhythmic risk. Reperfusion (by PCI or thrombolysis) reduces arrhythmic risk by restoring homogeneous conduction, though it can itself cause brief arrhythmia (reperfusion arrhythmia = AIVR). Hypokalaemia, hypomagnesaemia, and acidosis amplify arrhythmic risk — electrolyte correction is a critical supportive measure.
Conduction disturbance mechanism: The AV node is supplied by the AV nodal artery, a branch of the RCA in 90% of individuals. Inferior STEMI (RCA occlusion) can therefore interrupt AV nodal blood supply, causing Wenckebach (Mobitz I) or complete heart block. This is typically infra-nodal (His bundle level), generally resolves with reperfusion, and responds to atropine. In contrast, anterior STEMI involving the septum (proximal LAD) causes bilateral bundle branch block (RBBB + LAFB or LPFB) or complete heart block — this is a His-Purkinje level block, does NOT respond to atropine, and requires pacemaker insertion.
Mechanical complication mechanism: The necrotic myocardium undergoes two phases of structural change: early oedema and softening (hours 1–72), and then enzymatic autolysis by macrophage-mediated matrix metalloproteinases (days 3–5). The softened myocardium is mechanically vulnerable — it can rupture (free wall, septum) or fail to support the attached papillary muscle. The posteromedial papillary muscle is uniquely vulnerable because it receives its entire blood supply from a single vessel (the right posterior descending artery from the RCA), whereas the anterolateral papillary muscle has dual supply (LAD + LCx). This explains why papillary muscle rupture occurs predominantly in inferior STEMI (RCA territory) rather than anterior MI. The pressure gradient between the LV and the RV (or LA) determines the haemodynamic impact of VSD (L→R shunt, volume overload of the RV) and papillary muscle rupture (acute MR, massive LA pressure elevation and pulmonary oedema).
Dressler syndrome mechanism: Autoimmune pericarditis and pleuritis developing weeks after MI is caused by immune sensitisation to cardiac antigens (myosin, actin) released from the necrotic myocardium. T-lymphocyte activation and immune complex deposition in the pericardium produce the inflammatory response. The time course (2–8 weeks) is consistent with the adaptive immune response cycle. The same mechanism may occur weeks to months after cardiac surgery ('post-cardiac injury syndrome' or 'post-pericardiotomy syndrome').
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
Mechanical Complications of Acute Myocardial Infarction
Diagnosis and Investigation in Acute ACS Management
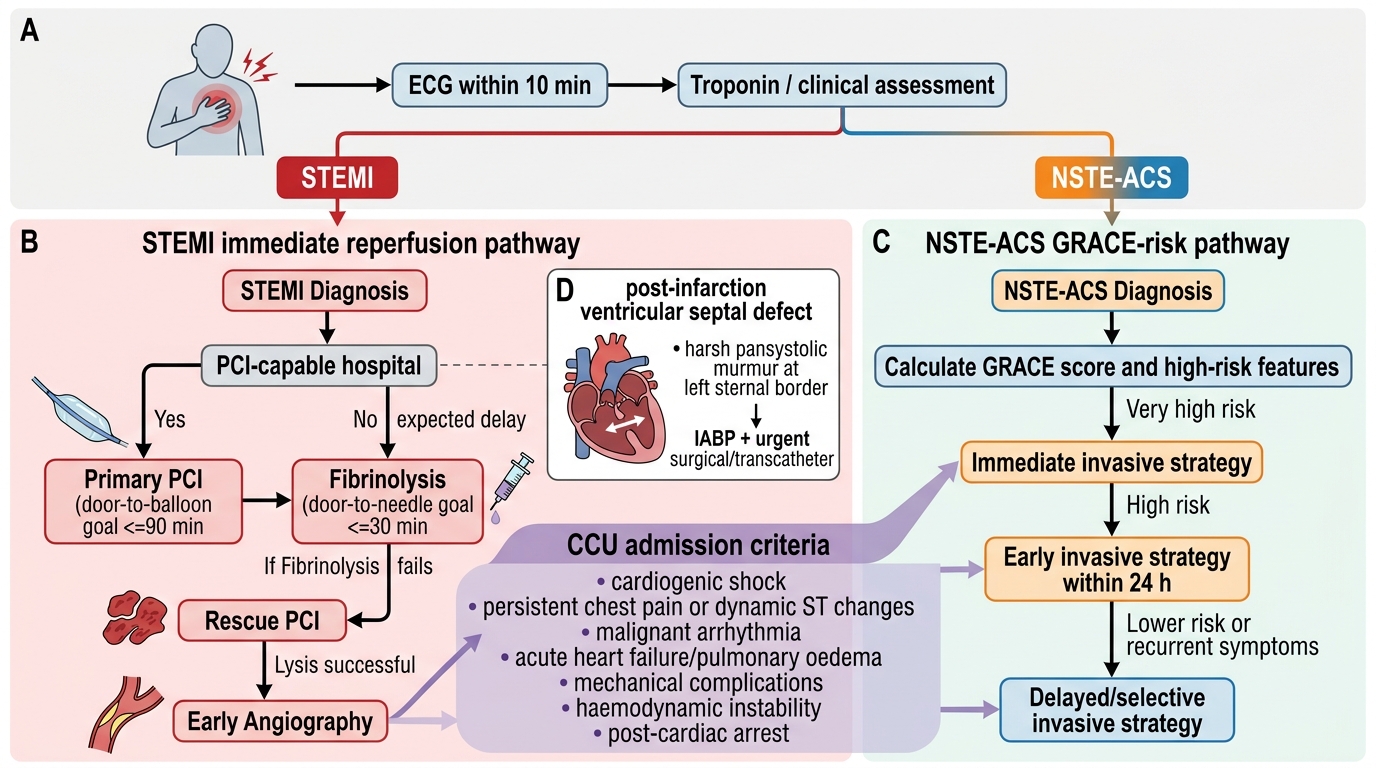
The diagnostic investigation in ACS acute management overlaps with the diagnostic module but focuses here on specific investigations that directly drive management decisions — distinguishing STEMI from NSTEMI (reperfusion vs risk-stratified invasive), identifying complications (echo, haemodynamic monitoring), and risk-stratifying severity (Killip class, GRACE score, TIMI score).
The CCU (Coronary Care Unit) admission criteria derive directly from these diagnostic assessments. A patient with suspected ACS requires CCU admission when any of the following are present: confirmed STEMI or high-risk NSTE-ACS (GRACE score >140), haemodynamic instability (Killip class II–IV, hypotension, tachycardia indicating low cardiac output), high-grade arrhythmia (VT, VF, CHB, new LBBB), or clinical need for continuous monitoring during the diagnostic phase. The CCU provides continuous cardiac monitoring, defibrillator availability, and immediate access to reperfusion therapy. Lower-risk patients (GRACE score <109, NSTE-ACS with negative troponin on serial testing) may be managed in a monitored step-down unit or observation ward.
The GRACE score is the validated risk stratification tool for NSTE-ACS and guides the timing of coronary angiography: GRACE >140 (high risk) → immediate invasive (angiography within 24 hours); GRACE 109–140 (intermediate) → early invasive (within 72 hours); GRACE <109 (low risk) → selective invasive (guided by stress imaging or non-invasive assessment). The TIMI risk score (7 variables: age ≥65, ≥3 CAD risk factors, prior stenosis ≥50%, ST deviation, ≥2 anginal events in 24h, aspirin use in past 7 days, elevated cardiac markers) is used at NSTE-ACS presentation: score 0–2 = low risk; 3–4 = intermediate; 5–7 = high risk.
Haemodynamic monitoring in cardiogenic shock (Killip IV): Continuous intra-arterial blood pressure monitoring (radial arterial line) provides real-time BP measurement more accurately than non-invasive cuffs in shock. Central venous pressure monitoring guides fluid management in RV infarction. Pulmonary artery catheterisation (Swan-Ganz catheter) may be used to measure pulmonary capillary wedge pressure (PCWP) — elevated PCWP (>18 mmHg) confirms LV failure (vs. volume depletion); very high PCWP (>25 mmHg) correlates with pulmonary oedema. However, routine Swan-Ganz use in ACS cardiogenic shock is not supported by randomised trials (ESCAPE trial) — clinical and echocardiographic assessment usually suffices.
Bedside echocardiography (point-of-care ultrasound, POCUS) provides rapid assessment of LVEF, regional wall motion, pericardial effusion, and mechanical complications in haemodynamically unstable ACS patients. A LVEF <30–35% in anterior STEMI indicates extremely high risk of cardiogenic shock and should prompt early escalation of support (inotropes, intra-aortic balloon pump or mechanical circulatory support).
ACS Management Pathway: STEMI vs NSTE-ACS
SELF-CHECK
A 63-year-old woman is admitted with anterior STEMI. On day 3, she develops sudden haemodynamic deterioration: BP 80/60 mmHg, HR 125/min, SpO2 85% on room air. Examination reveals a new harsh pansystolic murmur loudest at the left sternal border, with bilateral crepitations up to the mid-zones. Bedside echo shows a left-to-right shunt across the interventricular septum. What is the complication and the correct immediate management?
A. Acute mitral regurgitation from papillary muscle rupture — urgent mitral valve surgery
B. Post-infarction ventricular septal defect — stabilise with vasodilators if not in shock, intra-aortic balloon pump, and urgent surgical or transcatheter closure
C. Dressler syndrome pericarditis — start high-dose aspirin and colchicine
D. Reinfarction with cardiogenic shock — repeat ECG and retrigger the reperfusion pathway
Reveal Answer
Answer: B. Post-infarction ventricular septal defect — stabilise with vasodilators if not in shock, intra-aortic balloon pump, and urgent surgical or transcatheter closure
The new harsh pansystolic murmur at the left sternal border (not apex) with L→R shunt on echo colour Doppler confirms a post-infarction ventricular septal defect (VSD) — septal rupture from anterior STEMI affecting the LAD territory. This is a mechanical complication distinct from papillary muscle rupture (which produces a murmur at the apex with pulmonary oedema but no left-to-right shunt). Initial management: vasodilators (nitroprusside or nitroglycerine — reduce afterload and left-to-right shunt) if not in profound shock; intra-aortic balloon pump (IABP) for haemodynamic support by reducing afterload and augmenting diastolic pressure; urgent surgical patch closure or transcatheter VSD closure. Mortality without surgical repair approaches 90% in the first 2 weeks.