Page 1 of 24
IM2.1-5 | Atherosclerosis and IHD Foundations — SDL Guide
Learning Objectives
- Discuss the epidemiology and burden of ischaemic heart disease in India and globally
- Classify modifiable and non-modifiable risk factors for atherosclerosis
- Describe the lipid cycle including the role of LDL, HDL, and VLDL in atherogenesis
- Explain the pathogenesis and natural history of atherosclerotic plaque from fatty streak to complicated lesion
- Define and distinguish the acute coronary syndromes — STEMI, NSTEMI, and unstable angina
INSTRUCTIONS
Ischaemic heart disease is the single leading cause of death in India. This foundational module builds the conceptual architecture — epidemiology, risk stratification, lipid biology, plaque pathogenesis, and the spectrum of acute coronary syndromes — that underpins every clinical decision in the IM2 competency set. Work through each section in sequence; the self-assessment scenarios at the end integrate all five arc steps.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 295 — Ischaemic Heart Disease (textbook)
- API Textbook of Medicine, 10th ed., Ch. on Coronary Artery Disease (textbook)
- Davidson's Principles & Practice of Medicine, 24th ed., Ch. 18 — Cardiology (textbook)
- ACC/AHA 2022 Guideline for Coronary Artery Disease Management (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Ramesh is 48 years old, a non-smoker with well-controlled hypertension and a family history of 'heart attacks before 50'. He has never had chest pain. His fasting lipid profile reveals LDL-cholesterol 4.1 mmol/L and HDL 0.9 mmol/L. His cardiologist tells him he is at 'high cardiovascular risk' and recommends a statin even though he feels perfectly well. Ramesh is confused: why does he need a cardiac drug if his heart is functioning normally? The answer lies in understanding atherosclerosis as a decades-long biological process that is silently remodelling his coronary arteries long before any clinical event. Six months later, his colleague Suresh — 52, smoker, diabetic — arrives in the emergency department in diaphoresis with crushing chest pain radiating to the left jaw. His ECG shows 3-mm ST elevation in leads II, III, and aVF. Suresh is having an acute myocardial infarction. Both men's stories began at the same biological starting point: an atherosclerotic plaque forming in the wall of a coronary artery. This module explains how that plaque forms, why it ruptures, and how the spectrum of clinical syndromes from stable angina to STEMI represents different stages along a single pathological continuum.
WHY THIS MATTERS
Ischaemic heart disease is not merely a cardiology subspecialty topic — it is the single most common cause of death in India, accounting for over 28% of all adult deaths and disproportionately affecting men and women in the economically productive age group of 40–65 years. The Indian burden is distinctive: Indians develop IHD a decade earlier than Western populations, have a higher case-fatality rate for myocardial infarction, and present with more diffuse and complex coronary disease. The competency set IM2.1–2.5 requires you to understand epidemiology, risk factors, lipid pathophysiology, and the natural history of atherosclerosis at the knowledge-and-higher (KH) level — meaning you must be able to apply these concepts to clinical scenarios, counsel patients on risk, and explain the rationale for primary and secondary prevention to patients like Ramesh and Suresh.
RECALL
Activate your prior knowledge before proceeding. The coronary arteries — left anterior descending (LAD), left circumflex (LCx), and right coronary artery (RCA) — arise from the aortic root and supply distinct myocardial territories. Interruption of flow produces ischaemia in the corresponding territory, reflected on the ECG by ST changes and on biomarkers by troponin release. From your biochemistry foundation, recall that cholesterol is transported in the blood packaged as lipoproteins — particles of varying density (VLDL, LDL, IDL, HDL) differentiated by their apoprotein coat, lipid composition, and metabolic role. From your pharmacology training, recall that statins inhibit HMG-CoA reductase, the rate-limiting enzyme in hepatic cholesterol synthesis, thereby reducing LDL. The normal endothelium is anti-inflammatory, anti-thrombotic, and vasodilatory — its dysfunction is the trigger for atherogenesis. Keep these anchors in mind as you build the full mechanistic picture.
Epidemiology and Risk Factors of IHD
Ischaemic heart disease (IHD) encompasses the full spectrum of myocardial injury caused by imbalance between myocardial oxygen supply and demand, most often due to obstructive coronary artery disease (CAD) from atherosclerosis. In India, IHD has become the leading cause of death — estimated at 2.8 million deaths annually — driven by rapid urbanisation, dietary transition, and an epidemic of metabolic risk factors. Crucially, Indians develop IHD at a younger age than Europeans or Americans, with the peak incidence in the 45–64 age group compared to 55–74 in Western cohorts. The in-hospital case-fatality rate for STEMI in India remains 8–10%, higher than the 4–5% in developed countries, largely due to delays in presentation and limited access to reperfusion.
Globally, IHD remains the number one cause of death, accounting for approximately 9 million deaths per year (WHO 2023 estimates). The INTERHEART study — a landmark case-control study conducted in 52 countries including India — identified nine risk factors that together explain over 90% of the population-attributable risk for myocardial infarction. These nine factors are: smoking, dyslipidaemia (elevated ApoB/ApoA1 ratio), hypertension, diabetes mellitus, abdominal obesity, psychosocial stress, inadequate consumption of fruits and vegetables, physical inactivity, and alcohol consumption (protective at low doses, harmful at high). The INTERHEART data underscored that the modifiable risk factors far outweigh the non-modifiable ones in their contribution to preventable IHD.
Risk factors are conventionally classified as non-modifiable (cannot be changed by intervention) and modifiable (can be reduced by lifestyle or pharmacological means). This distinction has profound implications for both population-level public health strategy and individual clinical management.
Non-modifiable risk factors:
- Age: risk increases progressively with age; men ≥45 years and women ≥55 years (or post-menopausal) are in the higher risk category
- Sex: men have a higher risk than pre-menopausal women at any given age; the female risk catches up after menopause due to loss of oestrogen's cardioprotective effects
- Family history: first-degree relative with premature CAD — defined as MI or confirmed CAD in a male first-degree relative <55 years or female first-degree relative <65 years — confers a 1.5–2-fold increased risk; reflects inherited patterns of dyslipidaemia (e.g., familial hypercholesterolaemia), hypertension susceptibility, and other genetic determinants
- Ethnicity: South Asians have a 3–4-fold higher risk of premature CAD compared to Europeans; mechanisms include higher prevalence of insulin resistance, smaller lipoprotein particle size, and higher fibrinogen levels
Major modifiable risk factors:
- Smoking: the single most important modifiable risk factor; cigarette smoking increases CAD risk 2–4-fold by promoting endothelial dysfunction, increasing platelet aggregability, elevating fibrinogen, raising LDL, and lowering HDL; risk reduction begins within 1 year and approaches non-smoker levels by 5 years of cessation
- Dyslipidaemia: elevated LDL-cholesterol is the central atherogenic driver; high triglycerides (TG >150 mg/dL) and low HDL (<40 mg/dL in men, <50 mg/dL in women) independently increase risk; non-HDL cholesterol and ApoB are increasingly recognised as superior markers of atherogenic particle burden
- Hypertension: both systolic and diastolic hypertension increase CAD risk; the relationship is continuous, consistent, and independent — risk doubles for each 20/10 mmHg increment above 115/75 mmHg (Lewington meta-analysis)
- Diabetes mellitus: type 2 diabetes confers a 2–4-fold increased CAD risk; equivalent to the risk of a prior MI ('CAD risk equivalent'); associated with more diffuse coronary disease, more frequent silent ischaemia, and worse outcomes after MI
- Abdominal obesity: central adiposity (waist circumference >90 cm in Indian men, >80 cm in Indian women using Asian thresholds) drives insulin resistance and metabolic syndrome, increasing CAD risk independently of overall BMI
- Physical inactivity: sedentary lifestyle increases IHD risk by ~30–50%; regular aerobic exercise reduces risk by improving lipid profile, blood pressure, insulin sensitivity, and body weight
- Psychosocial factors: chronic psychological stress (workplace, socioeconomic), depression, and social isolation independently increase CAD risk; mechanisms include HPA axis activation, increased catecholamine levels, platelet hyper-reactivity, and adverse health behaviours
Emerging risk factors of relevance in India include elevated lipoprotein(a) [Lp(a)], high-sensitivity C-reactive protein (hsCRP as a marker of systemic inflammation), elevated homocysteine, and air pollution (particulate matter PM2.5 — increasingly linked to endothelial dysfunction and accelerated atherogenesis in Indian urban populations).
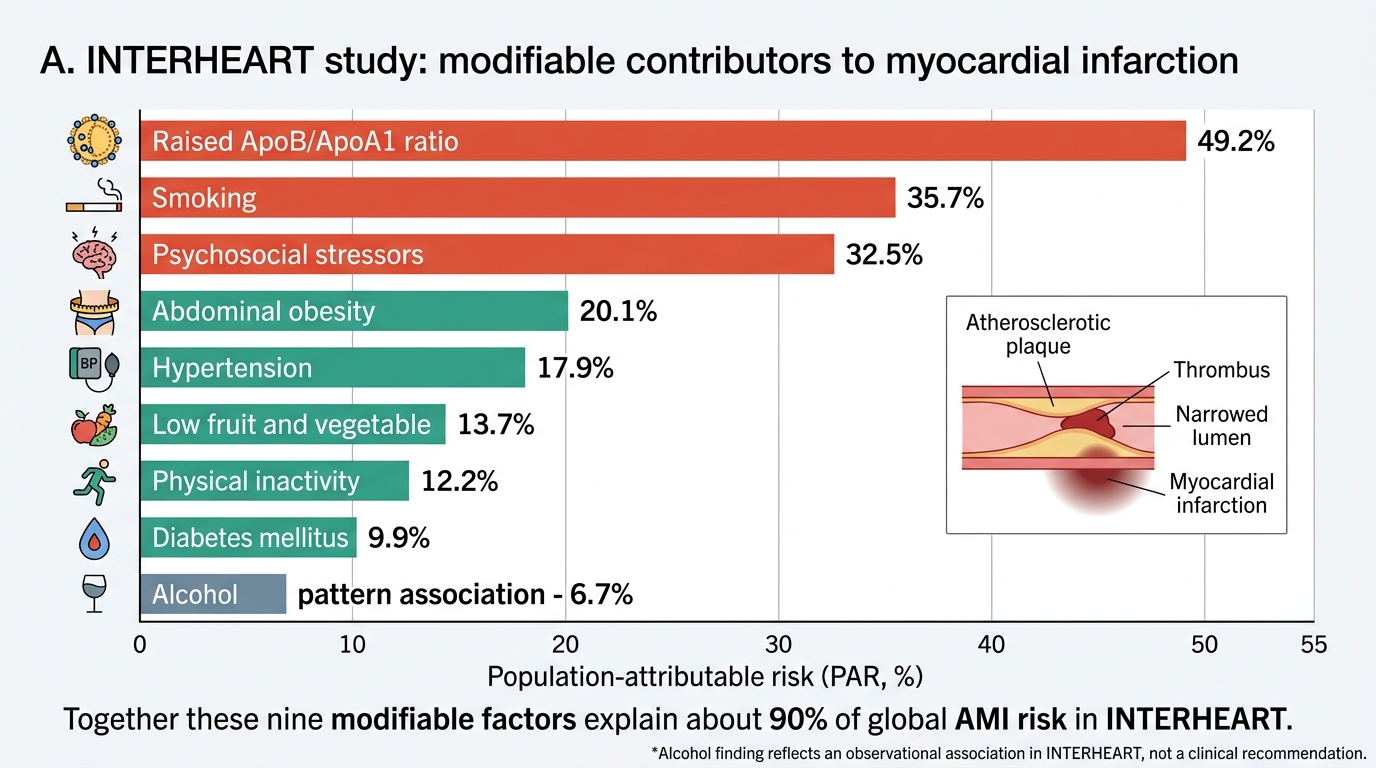
INTERHEART Modifiable Risk Factors for Myocardial Infarction
Lipid Cycle and the Role of Dyslipidaemia in Atherogenesis
Dyslipidaemia is both the most biologically central and the most actionable contributor to atherosclerosis. Understanding how lipids are transported and metabolised is essential for understanding why specific lipid fractions matter, why LDL-lowering is the cornerstone of prevention, and how to interpret a lipid profile in clinical context.
Cholesterol and triglycerides are hydrophobic molecules that cannot circulate freely in plasma; they must be transported packaged within lipoproteins — spherical particles with a hydrophobic lipid core and a hydrophilic outer shell of phospholipids and apolipoproteins (Apo). The apolipoproteins are not merely structural — they are metabolic determinants: they direct lipoproteins to specific receptors and serve as cofactors for enzymes that remodel the particles.
The exogenous (dietary) pathway: Dietary fat and cholesterol absorbed in the small intestine are packaged into chylomicrons in enterocytes, with ApoB-48 as the primary structural apoprotein. Chylomicrons enter the lymphatics, reach the bloodstream via the thoracic duct, and deliver triglycerides to peripheral tissues (muscle, adipose) via the enzyme lipoprotein lipase (LPL). After triglyceride removal, the chylomicron remnant — enriched in cholesterol esters — is taken up by the liver via ApoE-mediated receptor binding.
The endogenous pathway: The liver synthesises VLDL (very low density lipoprotein), which carries triglycerides to peripheral tissues. LPL progressively removes triglyceride from VLDL, converting it first to IDL (intermediate density lipoprotein) and then to LDL (low density lipoprotein). LDL is the terminal product — a cholesterol-rich, triglyceride-poor particle bearing ApoB-100 as its single structural apoprotein. LDL is cleared from the circulation by LDL receptors (LDLR) on hepatocytes and peripheral cells; receptor expression is upregulated by statins (via HMG-CoA reductase inhibition → reduced intracellular cholesterol → LDLR upregulation). Familial hypercholesterolaemia (FH) results from LDLR mutations that impair LDL clearance, producing dramatically elevated LDL and premature CAD.
The reverse cholesterol transport pathway: HDL (high density lipoprotein), synthesised in the liver and intestine as lipid-poor nascent particles bearing ApoA-I, extracts cholesterol from peripheral tissues — including foam cells in atherosclerotic plaques — via the transporter ABCA1. The cholesterol-enriched HDL then delivers its cargo to the liver (via SR-BI receptors) or transfers cholesterol esters to LDL via CETP (cholesteryl ester transfer protein) in exchange for triglycerides. This reverse transport is cardioprotective — HDL 'de-loads' the plaque. Low HDL therefore indicates impaired reverse transport capacity, and high triglycerides often accompany low HDL because of CETP-mediated exchange.
Atherogenic dyslipidaemia — the pattern characteristic of metabolic syndrome and type 2 diabetes in India — comprises the triad of elevated triglycerides, low HDL, and small dense LDL particles. Small dense LDL particles are more atherogenic per particle than large buoyant LDL because they bind LDL receptors less avidly (longer plasma half-life), penetrate the arterial intima more readily, and are more susceptible to oxidative modification. Measuring non-HDL cholesterol (= total cholesterol − HDL; target <130 mg/dL in high-risk patients) or ApoB (target <80 mg/dL in high-risk) captures this atherogenic particle burden better than LDL-cholesterol alone.
Lipoprotein(a) [Lp(a)] is a modified LDL with an additional apoprotein — apo(a) — linked to ApoB-100 by a disulphide bond. High Lp(a) (>50 mg/dL) independently increases CAD risk by promoting both atherogenesis (enters the arterial wall preferentially) and thrombosis (apo(a) is structurally homologous to plasminogen and inhibits fibrinolysis). Lp(a) is genetically determined (minimally affected by diet or exercise) and is not reduced by statins — an important clinical fact.
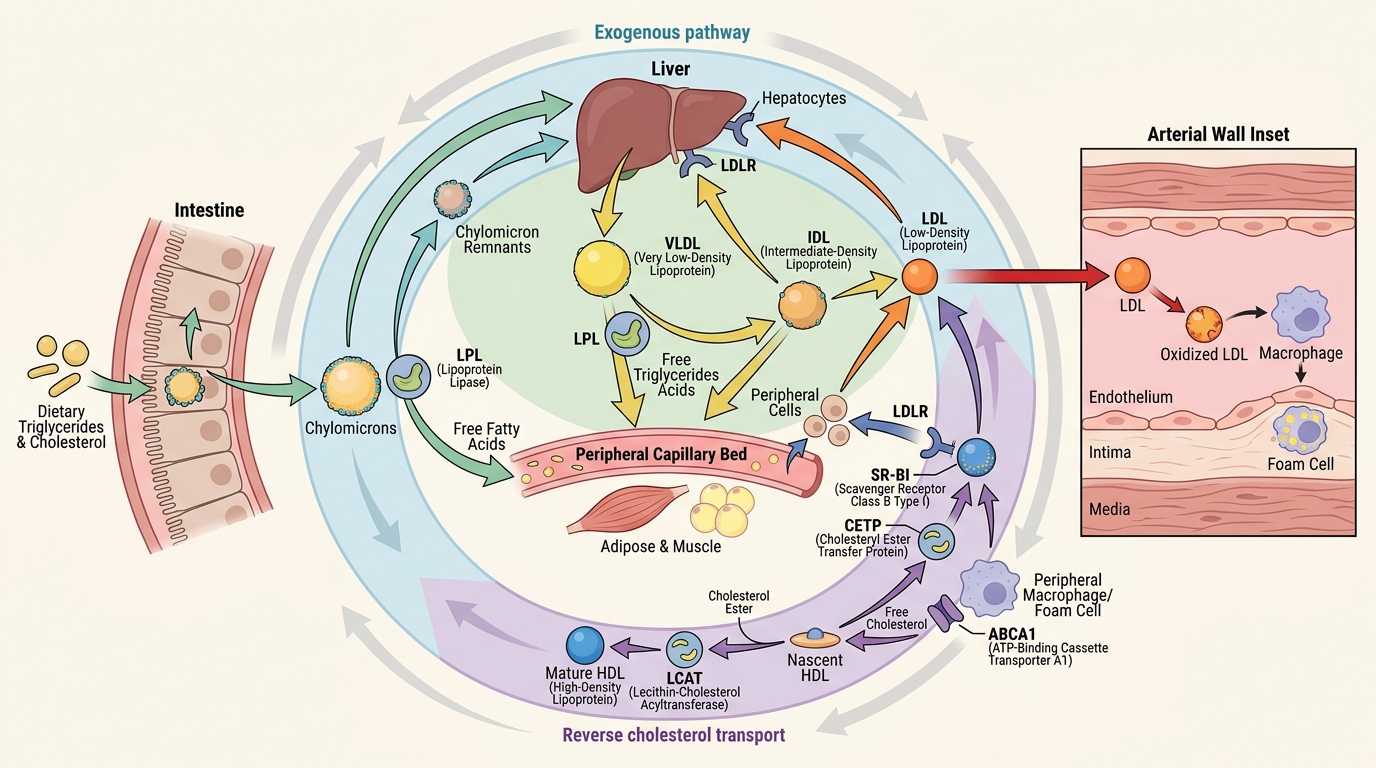
Lipid Transport Pathways and LDL Entry into the Arterial Wall
Pathogenesis and Natural History of Atherosclerosis
Atherosclerosis is a chronic inflammatory disease of large and medium-sized arteries characterised by the subintimal accumulation of lipid, inflammatory cells, smooth muscle cells, and fibrous matrix. The response-to-injury hypothesis — first articulated by Ross (1976, refined 1999) — frames the entire process: atherosclerosis begins with endothelial injury or dysfunction, progresses through inflammatory cell recruitment and lipid accumulation, and culminates in a complicated plaque that may rupture, triggering arterial thrombosis and acute ischaemia.
Step 1 — Endothelial dysfunction: The normal endothelium maintains vascular homeostasis through production of nitric oxide (vasodilation), prostacyclin (anti-platelet), thrombomodulin (anti-coagulant), and tissue plasminogen activator (fibrinolytic). Endothelial dysfunction — triggered by haemodynamic shear stress (particularly at arterial bifurcations where turbulent flow occurs), oxidised LDL (oxLDL), hyperglycaemia, nicotine, and hypertension — converts the endothelium from this protective phenotype to a pro-inflammatory, pro-adhesive one. The dysfunctional endothelium upregulates expression of adhesion molecules — VCAM-1 (vascular cell adhesion molecule-1), ICAM-1, and selectins — which tether circulating monocytes to the endothelial surface. This is the earliest detectable event in atherogenesis and occurs before any grossly visible lesion.
Step 2 — Fatty streak formation: Monocytes adhere to the dysfunctional endothelium, transmigrate into the subintimal space, and differentiate into macrophages under the influence of M-CSF and GM-CSF. In the subintima, macrophages encounter LDL particles — particularly the oxidatively modified oxLDL — and engulf them via scavenger receptors (SR-A, CD36). Unlike the LDLR, scavenger receptors are not downregulated by intracellular cholesterol accumulation, so macrophages continue to ingest lipid until they become bloated with cholesterol ester droplets — transforming into foam cells. Accumulations of foam cells and lipid-laden smooth muscle cells just beneath the endothelium form the fatty streak — the earliest visible lesion in the arterial intima. Fatty streaks are present in the aortas of children as young as 10 years and in the coronary arteries of young adults in their twenties. They are fully reversible at this stage.
Step 3 — Intermediate plaque (fibrous plaque): Foam cells and activated macrophages secrete cytokines (IL-1β, TNF-α, IL-6) and growth factors (PDGF, FGF) that recruit vascular smooth muscle cells (VSMCs) from the media, stimulating their proliferation and migration into the intima. VSMCs produce collagen, elastin, and proteoglycans that form the fibrous cap over the growing lipid core. The plaque now has a three-part architecture: a central necrotic lipid core (containing foam cell debris, free cholesterol crystals, and apoptotic cells), a surrounding fibrous cap of VSMCs and collagen, and an adventitial shoulder region highly infiltrated with macrophages. This is the vulnerable area.
Step 4 — Plaque progression and complications: Advanced plaques cause luminal narrowing (stenosis) as they expand. However, early plaques often expand outward (positive remodelling — Glagov phenomenon) without narrowing the lumen, which means a haemodynamically significant plaque on angiography does not necessarily predict which plaque will rupture next. The critical event in ACS is plaque rupture or erosion:
- Plaque rupture: the fibrous cap is thinned by matrix metalloproteinases (MMPs) secreted by activated macrophages. A thin-cap fibroatheroma (TCFA) — cap thickness <65 µm — is the 'vulnerable plaque'. Rupture exposes the highly thrombogenic lipid core to the flowing blood, triggering platelet adhesion, activation, and aggregation via collagen/vWF–GPIb and thromboxane A2, followed by the coagulation cascade → acute thrombus.
- Plaque erosion (more common in younger women and diabetics): thrombus forms on a denuded endothelial surface without fibrous cap rupture; the underlying plaque may be less lipid-rich.
The thrombus may be: (1) occlusive → STEMI; (2) partially occlusive → NSTEMI (troponin elevation without ST elevation); (3) transiently occlusive → unstable angina (no permanent myocyte necrosis, no troponin rise). The time-course of myocardial necrosis follows the wavefront phenomenon (Reimer & Jennings, 1977): transmural necrosis proceeds from endocardium to epicardium over 3–6 hours. This biological clock underpins the 'time is muscle' principle — the faster reperfusion is achieved, the greater the myocardium salvage.
Chronic stable angina by contrast reflects a haemodynamically significant stenosis (typically >70% diameter reduction) that limits coronary flow reserve sufficiently to produce ischaemia during exertion but does not result in acute thrombosis. The plaque is usually heavily calcified, with a thick fibrous cap — mechanically stable, not acutely vulnerable.
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
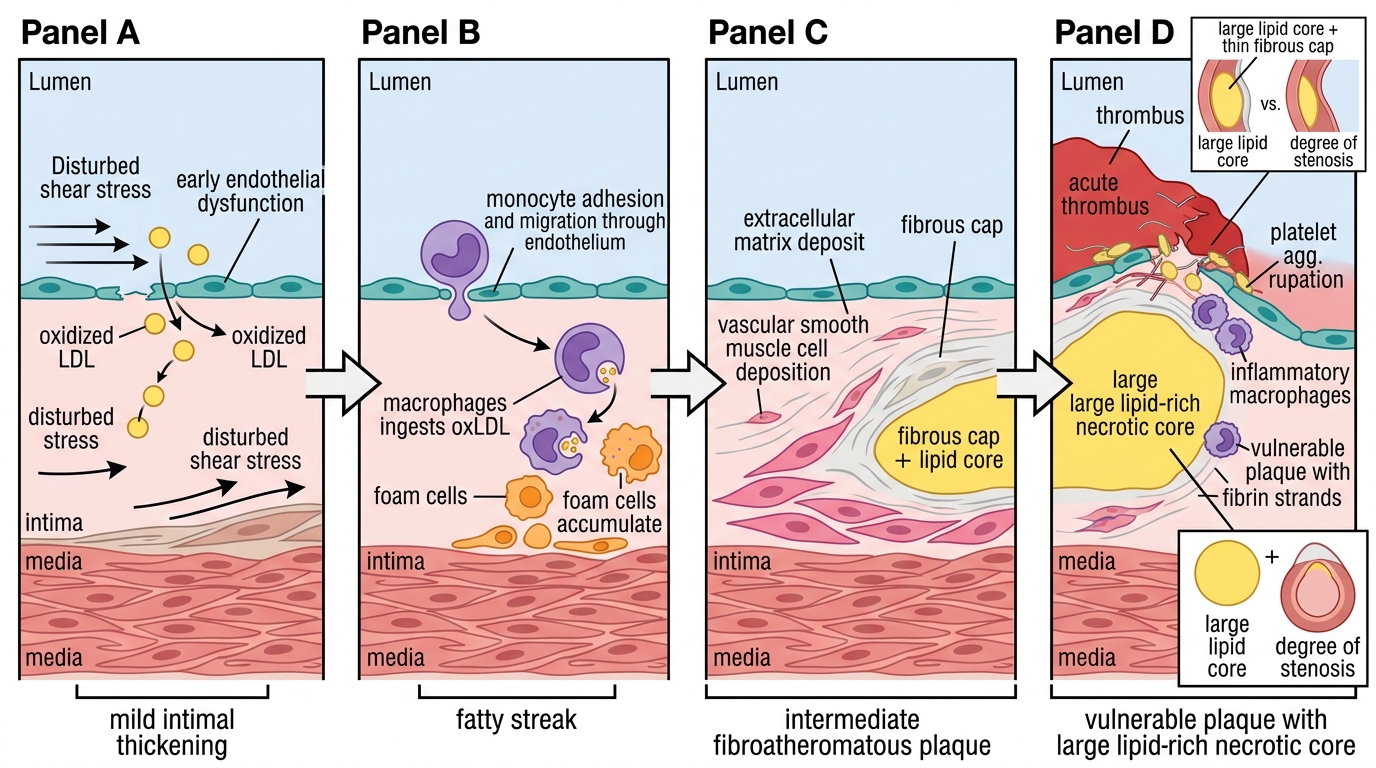
Pathogenesis of Atherosclerotic Plaque Rupture
SELF-CHECK
A 55-year-old man with type 2 diabetes, hypertension, and a 20-pack-year smoking history undergoes an exercise stress test that is strongly positive. Coronary CT angiography shows 50% stenosis of the LAD with a large lipid-rich core and a thin fibrous cap. The MOST accurate statement about this lesion is:
A. Because it is only 50% stenosis, it is unlikely to cause an acute coronary event
B. This is a stable, calcified plaque with low rupture risk
C. This thin-cap fibroatheroma is at high risk of rupture and may cause an ACS without prior warning symptoms
D. Atherosclerosis this advanced is irreversible and will not respond to statin therapy
Reveal Answer
Answer: C. This thin-cap fibroatheroma is at high risk of rupture and may cause an ACS without prior warning symptoms
Plaque vulnerability depends on composition (lipid core size, fibrous cap thickness) rather than degree of stenosis. A thin-cap fibroatheroma (cap <65 µm) with a large necrotic core is a 'vulnerable plaque' highly prone to rupture regardless of the degree of luminal stenosis. In fact, >60% of acute MIs originate from plaques that were causing <50% stenosis before the event — because such plaques are often lipid-rich and have not yet developed the calcification that stabilises them. Statins do reduce plaque vulnerability by reducing lipid content and inflammation, even if they cannot fully reverse advanced lesions.