Page 11 of 17
IM23.4-7 | Sodium and Potassium Disorders — SDL Guide (Part 2)
Hypernatraemia: Causes and Management
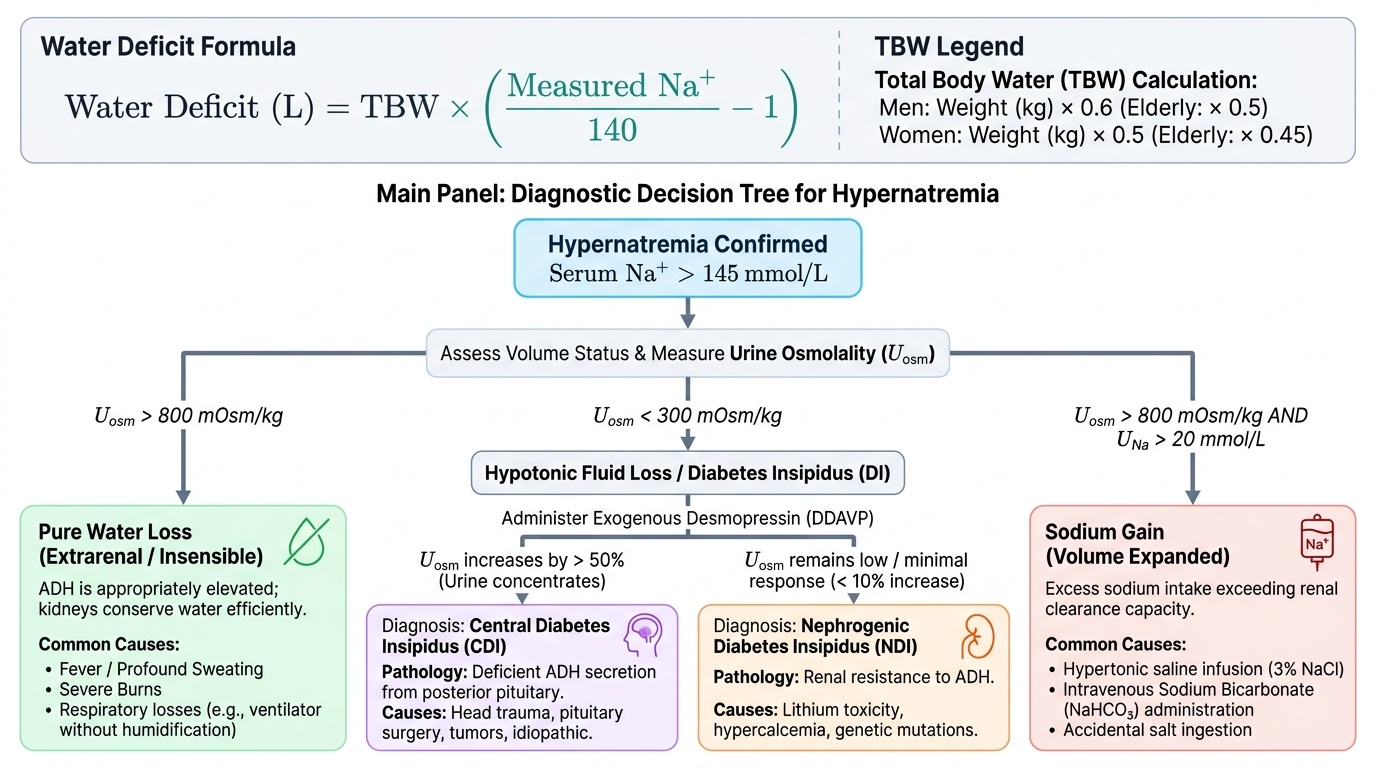
Hypernatraemia is defined as serum sodium >145 mmol/L and always indicates a hyperosmolar state — free water deficit relative to sodium. Unlike hyponatraemia, hypernatraemia cannot exist in a conscious patient who has access to water and an intact thirst mechanism, because thirst would drive the individual to drink and correct it. Hypernatraemia therefore occurs predominantly in patients who cannot access or communicate their thirst: the elderly with impaired consciousness, ICU patients, neonates, and patients with impaired hypothalamic thirst sensation (hypodipsic hypernatraemia). It can also occur in states where water losses are so rapid that even an intact thirst response cannot compensate — for example, in severe burns, high fevers, or profound osmotic diuresis. The three broad mechanisms are pure water loss (no solute), hypotonic fluid loss where water is lost in excess of sodium (as in diabetes insipidus or osmotic diuresis), and rarely sodium gain from iatrogenic sources. In each case the treatment is restitution of free water using hypotonic fluids, but the rate of correction must be controlled in the chronic setting because rapid correction causes cerebral oedema in a brain that has accumulated osmolytes to adapt to the hyperosmolar state. The key diagnostic test that distinguishes the two forms of diabetes insipidus — central versus nephrogenic — is the response to exogenous desmopressin (DDAVP): urine concentrates in central DI (ADH is missing and replaced) but remains dilute in nephrogenic DI (the kidney cannot respond).
Provided image
The causes of hypernatraemia are classified by mechanism: — classified by mechanism:
- Pure water loss (increased insensible losses): fever, burns, respiratory losses during mechanical ventilation (humidifier-free circuits), sweating. ADH is appropriately maximally elevated; kidneys produce maximally concentrated urine (>800 mOsm/kg). Urine osmolality appropriately high.
2. Hypotonic fluid loss (water loss exceeding sodium loss):
- Central diabetes insipidus (CDI): deficient ADH secretion from posterior pituitary damage (trauma, surgery, pituitary mass, Langerhans cell histiocytosis, autoimmune, idiopathic). Urine is inappropriately dilute despite hypernatraemia (urine osmolality <300 mOsm/kg, often <100). Responds to desmopressin (DDAVP) — synthetic V2 agonist.
- Nephrogenic diabetes insipidus (NDI): kidneys fail to respond to ADH. Causes: hypercalcaemia (calcium impairs aquaporin-2 insertion), hypokalaemia, lithium (blocks V2 receptor signalling), demeclocycline, severe CKD. Urine is dilute; does NOT respond to desmopressin. Treatment: low-sodium low-protein diet, thiazide diuretics (paradoxically reduce polyuria by reducing tubular delivery to the collecting duct).
- Osmotic diuresis: glucosuria (uncontrolled diabetes mellitus), mannitol infusion, high-protein feeds — non-reabsorbable solutes drag water into urine, causing both hypernatraemia and polyuria with moderately concentrated urine.
- Sodium gain (uncommon): iatrogenic — hypertonic saline, sodium bicarbonate infusion, hypertonic feeding formulas. Seawater near-drowning.
Water deficit calculation: the free water deficit in litres = TBW × [(serum Na / 140) − 1], where TBW = 0.6 × weight (kg) in men, 0.5 × weight in women.
Management of hypernatraemia: replace the free water deficit with hypotonic fluids (5% dextrose, 0.45% saline, or oral water). The rate of correction must also be controlled: do NOT correct faster than 0.5–1 mmol/L/hour (maximum 10–12 mmol/L in 24 hours) in chronic hypernatraemia (>48 hours' duration) — too-rapid correction causes cerebral oedema as brain cells that have accumulated osmolytes to compensate now swell. Account for ongoing insensible losses in the replacement calculation. Diabetes insipidus is treated with desmopressin (CDI: intranasal or IV/SC; NDI: thiazides ± amiloride for lithium-induced NDI).
Hypokalaemia: Causes, Clinical Features, and Management
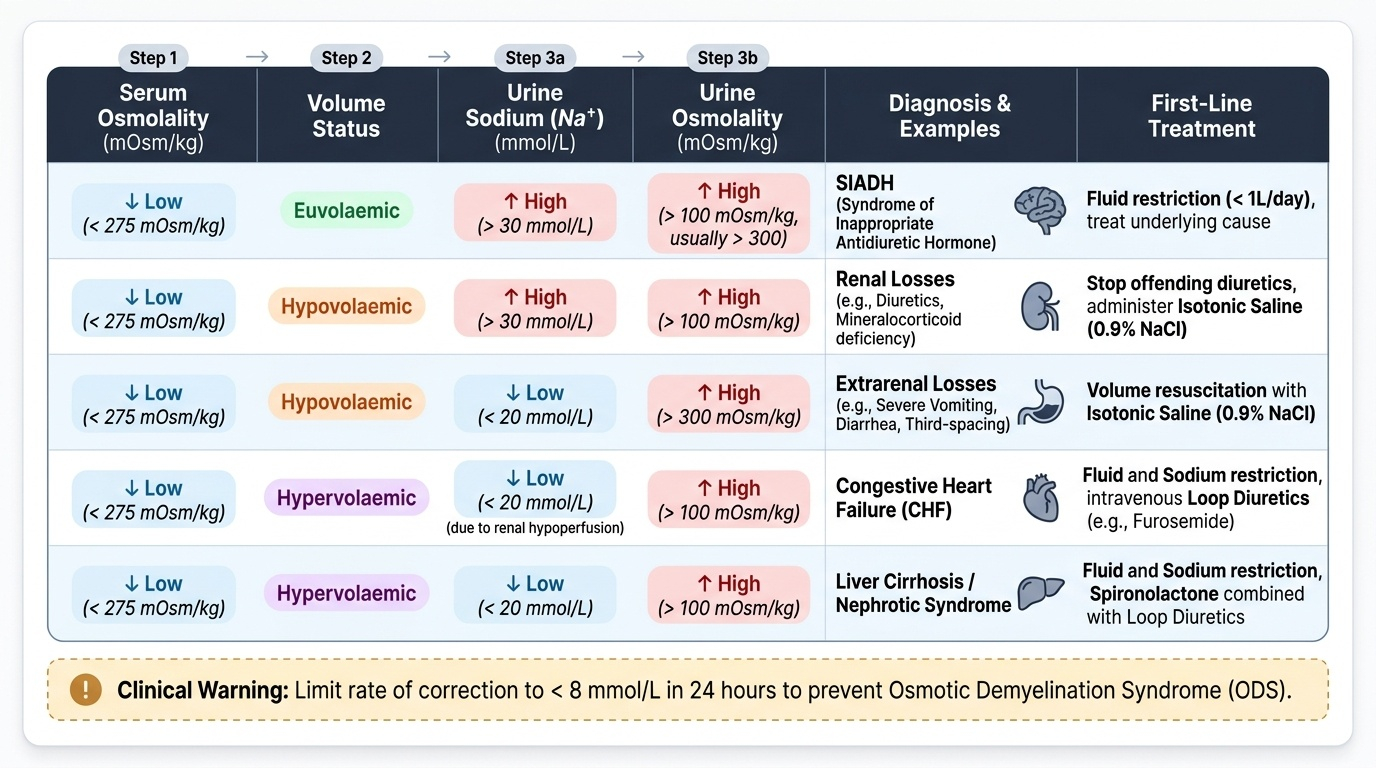
Hypokalaemia is defined as serum potassium <3.5 mmol/L. It is extremely common in hospitalised patients, particularly those receiving diuretics, nasogastric suction, or prolonged intravenous fluids without potassium supplementation. The clinical significance of hypokalaemia arises from its effects on cardiac, skeletal muscle, and renal function. Before evaluating causes, recall that serum potassium reflects only 2% of total body potassium — the rest is intracellular. A serum K⁺ of 3.0 mmol/L may represent a total body deficit of 200–400 mmol, while a serum K⁺ of 2.5 mmol/L may indicate a deficit of 400–600 mmol. This non-linear relationship means that the serum level underestimates the severity of depletion at low values, and aggressive replacement is sometimes required. The diagnostic distinction between renal and extrarenal potassium loss is made by measuring urine potassium: a urine K⁺ above 20 mmol/L (or a spot urine K-to-creatinine ratio >20 mmol/mmol) in a hypokalaemic patient means the kidney is wasting potassium inappropriately — suggesting diuretic use, hyperaldosteronism, hypomagnesaemia, or a tubular disorder. A urine K⁺ below 20 mmol/L means the kidney is conserving potassium correctly and the losses are occurring elsewhere — typically the gastrointestinal tract or through inadequate intake.
The causes of hypokalaemia are classified by mechanism: — classified by mechanism:
1. Transcellular shift (K⁺ moves from ECF into cells):
- Insulin excess (exogenous or endogenous): activates Na⁺/K⁺-ATPase → K into cells. Explains why diabetic ketoacidosis treatment (insulin infusion) can cause rapid hypokalaemia even in a patient who was initially hyperkalaemic — total body K⁺ was already depleted (urinary K losses from osmotic diuresis), and insulin accelerates the shift.
- Catecholamines (β₂ adrenergic stimulation — stress, salbutamol, adrenaline): Na⁺/K⁺-ATPase activation.
- Alkalosis: H⁺ shifts out of cells (to buffer the extracellular alkalosis) in exchange for K⁺ moving in; for every 0.1 unit rise in pH, K⁺ falls approximately 0.5–0.6 mmol/L.
- Periodic paralysis: hypokalaemic periodic paralysis (familial or thyrotoxic) — episodic attacks of weakness with marked hypokalemia from sudden transcellular shifts.
2. Renal losses (urine K⁺ >20 mmol/L or TTKG >4):
- Diuretics (most common cause in hospital): thiazides and loop diuretics (frusemide) increase distal tubular sodium delivery → aldosterone-driven K⁺ secretion.
- Primary hyperaldosteronism (Conn syndrome): autonomous aldosterone secretion by an adrenal adenoma or bilateral hyperplasia. Hallmarks: hypokalaemia + hypertension + metabolic alkalosis. Aldosterone-to-renin ratio >30 (with elevated aldosterone >15 ng/dL) is the screening test.
- Secondary hyperaldosteronism: elevated angiotensin II from renal artery stenosis, malignant hypertension, renin-secreting tumour.
- Magnesium deficiency: Mg²⁺ is required to keep ROMK channels closed (preventing K⁺ leak). Hypomagnesaemia causes renal K⁺ wasting that is refractory to K⁺ replacement alone — always check and correct Mg²⁺ when replacing K⁺.
- Bartter syndrome / Gitelman syndrome: rare genetic channelopathies of tubular transporters causing severe renal K and Na wasting; present in young patients with severe hypokalaemia and metabolic alkalosis without hypertension.
- RTA type 1 and type 2, aminoglycosides, amphotericin B, cisplatin.
3. Extrarenal losses (urine K⁺ <20 mmol/L):
- Gastrointestinal losses: diarrhoea (direct K⁺ loss), vomiting (secondary — vomit contains little K⁺; the alkalosis from HCl loss causes renal K⁺ wasting).
- Inadequate intake: rare as sole cause but contributes in anorexia, alcoholism, prolonged fasting.
Clinical features: at K⁺ 3.0–3.5 mmol/L — mild muscle weakness, fatigue, constipation. At K⁺ 2.5–3.0 mmol/L — significant muscle weakness, cramps, myalgia. At K⁺ <2.5 mmol/L — severe proximal myopathy (difficulty rising from chair, climbing stairs), ileus, respiratory muscle involvement, rhabdomyolysis. Cardiac: hypokalaemia prolongs the QT interval and predisposes to potentially fatal arrhythmias — ventricular tachycardia, torsades de pointes (especially in patients on digoxin — hypokalaemia potentiates digoxin toxicity by increasing binding affinity for Na⁺/K⁺-ATPase), atrial fibrillation. ECG changes: U waves (best seen in V2–V3), flattened or inverted T waves, T-U fusion, prolonged QU interval.
Management: correct the underlying cause (stop causative diuretic, treat diarrhoea). Replace potassium: oral KCl is preferred if the patient can tolerate it and is not severely symptomatic. IV KCl is given for severe (K⁺ <2.5 mmol/L) or symptomatic hypokalaemia, cardiac arrhythmias, or inability to take oral route. Maximum safe IV rate: 10–20 mmol/hour via peripheral line; up to 40 mmol/hour via central line with cardiac monitoring. Concentration: maximum 40 mmol/L via peripheral vein; higher concentrations cause phlebitis. Always check and correct magnesium concurrently — hypokalaemia refractory to K⁺ replacement alone strongly suggests hypomagnesaemia.
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
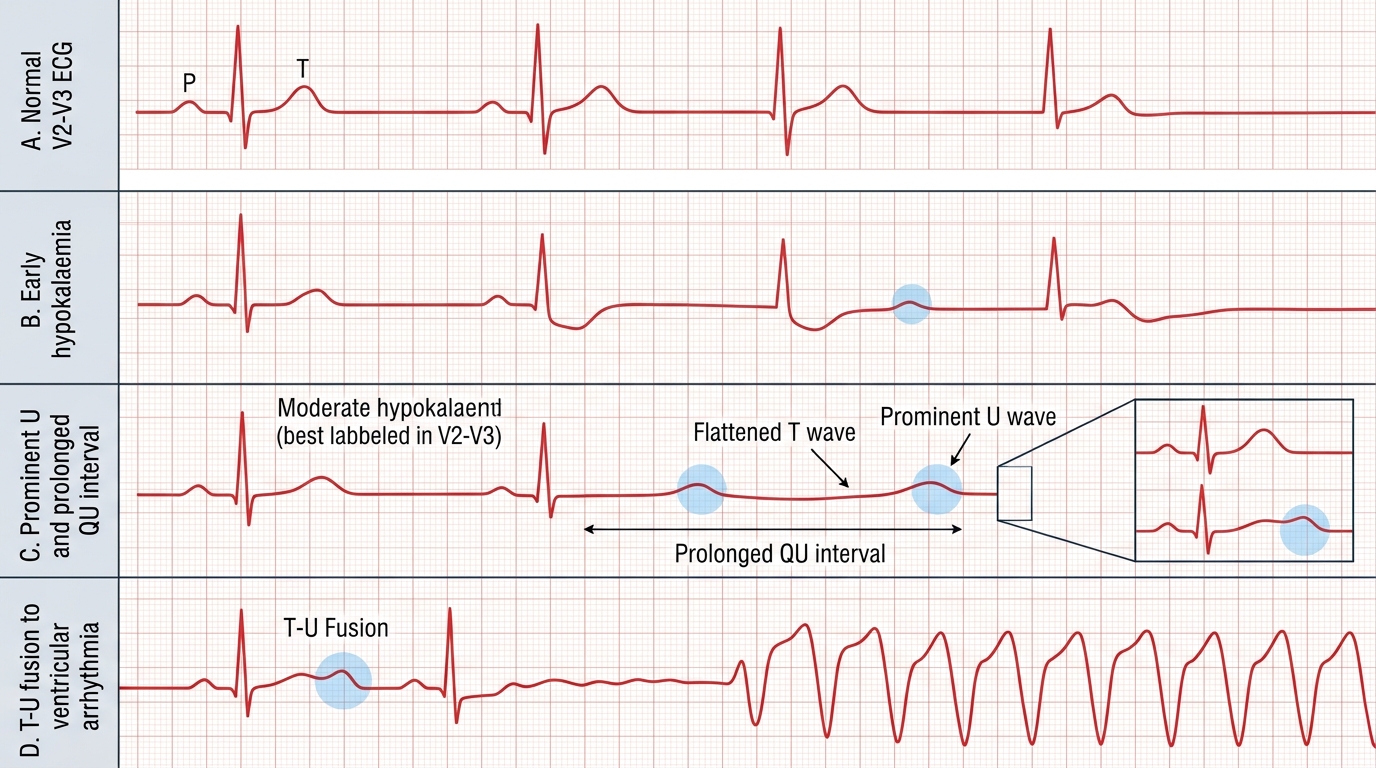
ECG Changes in Hypokalaemia
Hyperkalaemia: Causes, ECG Features, and Emergency Management
Hyperkalaemia is defined as serum potassium >5.0 mmol/L. It is a potentially immediately lethal disorder because of its cardiac effects, and every physician must be able to recognise the ECG sequence of hyperkalaemia and institute treatment in the correct order before even waiting for a repeat potassium confirmation. A serum K⁺ of 7.0 mmol/L with absent P waves on ECG is a cardiac emergency; a K⁺ of 5.8 mmol/L with a normal ECG in a patient with stable CKD is a chronic management problem. These are clinically different situations requiring different urgency of response, and the ECG is the arbiter. The pathophysiology of the cardiac toxicity of hyperkalaemia is a progressive reduction in the resting membrane potential: as extracellular K⁺ rises, the ratio of intracellular to extracellular K⁺ falls, and the resting potential becomes less negative (partial depolarisation). Initially this increases cardiac excitability — producing the peaked T waves of early hyperkalaemia. As depolarisation becomes more profound, sodium channels are inactivated and the cardiac conduction system fails, leading to progressive slowing — PR prolongation, P wave loss, QRS widening — and ultimately ventricular fibrillation or asystole. Understanding this progression allows the physician to interpret each ECG finding as a step on a predictable physiological ladder and to respond proportionally.
The causes of hyperkalaemia are classified by mechanism:
- Spurious hyperkalaemia (pseudohyperkalaemia — most common 'cause' on a routine panel): haemolysis from traumatic venepuncture, prolonged time to processing (K⁺ leaks from red cells at room temperature), very high white cell count (CML) or platelet count (essential thrombocythaemia) releasing intracellular K⁺ during clotting. Always repeat with a carefully drawn, promptly processed sample before treating a non-urgent result.
- Reduced renal excretion (most common true cause): chronic kidney disease (eGFR <30 mL/min — impaired tubular K⁺ secretion), acute kidney injury, hypoaldosteronism (Addison disease, hyporeninaemic hypoaldosteronism = type 4 renal tubular acidosis — commonest form, seen in diabetic nephropathy), drugs — ACE inhibitors, ARBs (block angiotensin II → reduce aldosterone), potassium-sparing diuretics (spironolactone, eplerenone, amiloride, triamterene), NSAIDs (reduce renin release), trimethoprim (blocks ROMK channel in collecting duct), heparin (inhibits aldosterone synthesis), calcineurin inhibitors (tacrolimus, ciclosporin).
3. Transcellular shift (K⁺ from cells into ECF):
- Acidosis (metabolic or respiratory): for every 0.1 unit fall in pH, K⁺ rises approximately 0.5–0.6 mmol/L. Particularly pronounced with inorganic acidoses (HCl, renal tubular acidosis) — less marked with organic acidoses (lactic acidosis, ketoacidosis).
- Insulin deficiency (diabetic ketoacidosis): despite total body K⁺ depletion, serum K⁺ is elevated at presentation due to the shift; K⁺ falls rapidly with insulin therapy.
- β-blockade (blocks catecholamine-driven K⁺ uptake into cells).
- Hyperosmolality (solvent drag of water out of cells carries K⁺ with it).
- Rhabdomyolysis, massive haemolysis, tumour lysis syndrome: massive cell destruction releases intracellular K⁺.
- Succinylcholine (depolarising neuromuscular blocker): opens K⁺ channels, releasing K⁺ — particularly dangerous in burns, crush injury, prolonged immobilisation, where upregulated acetylcholine receptors amplify the effect — can cause fatal cardiac arrest; contraindicated in these conditions.
- Excessive intake (unusual as sole cause): K⁺ supplements with impaired renal function, potassium-containing salt substitutes (KCl) used in dietary salt restriction.
ECG sequence of hyperkalaemia — memorise this as a progression, not isolated findings:
| K⁺ level | ECG change |
|---|---|
| 5.5–6.5 mmol/L | Tall peaked (tented) T waves — narrow base, symmetric, high amplitude; best in precordial leads |
| 6.5–7.0 mmol/L | Prolonged PR interval, flattening or loss of P waves |
| 7.0–8.0 mmol/L | Wide QRS complex (conduction delay) |
| >8.0 mmol/L | Sine wave pattern (P waves absent, wide QRS merging with T wave), ventricular fibrillation, asystole |
Emergency management of hyperkalaemia — sequence matters critically:
The management follows three distinct physiological phases: membrane stabilisation, transcellular shift, and total body removal. These are not interchangeable — stabilising the membrane is the most urgent step and must precede removal.
- Membrane stabilisation (acts in 1–3 minutes): Intravenous calcium gluconate 10 mL of 10% solution (or calcium chloride 5–10 mL of 10%) over 3–5 minutes. Calcium raises the threshold for cardiac membrane depolarisation, protecting against arrhythmias. It does NOT lower serum K⁺. Repeat if ECG changes persist or recur. Caution: do not give calcium if the patient is on digoxin (calcium potentiates digoxin toxicity — 'stone heart') unless the hyperkalaemia is immediately life-threatening.
2. Transcellular shift (acts in 15–30 minutes, lowers K⁺ by 0.5–1.5 mmol/L):
- Insulin + glucose: 10 units actrapid IV with 25–50 g glucose (50 mL of 50% dextrose); drives K⁺ into cells. Monitor glucose every 30–60 minutes for hypoglycaemia.
- Salbutamol nebulised 10–20 mg (high dose): β₂ agonist stimulates Na⁺/K⁺-ATPase. Acts within 15–30 minutes; lowers K⁺ by 0.5–1 mmol/L additive to insulin. May cause tachycardia.
- Sodium bicarbonate (only in metabolic acidosis context): 50–100 mmol IV sodium bicarbonate in metabolic acidosis corrects pH and promotes K⁺ shift. Less effective as sole agent in non-acidotic hyperkalaemia.
3. Total body removal (acts over hours to days):
- Loop diuretics (frusemide 40–80 mg IV) if renal function is adequate: promote urinary K⁺ excretion.
- Potassium-binding resins: sodium polystyrene sulphonate (Kayexalate) — onset hours; efficacy contested in acute setting; constipation risk. Patiromer or sodium zirconium cyclosilicate — newer, better-tolerated options.
- Haemodialysis or haemofiltration: definitive for severe or refractory hyperkalaemia, especially in acute kidney injury or end-stage renal failure. Most efficient removal method.
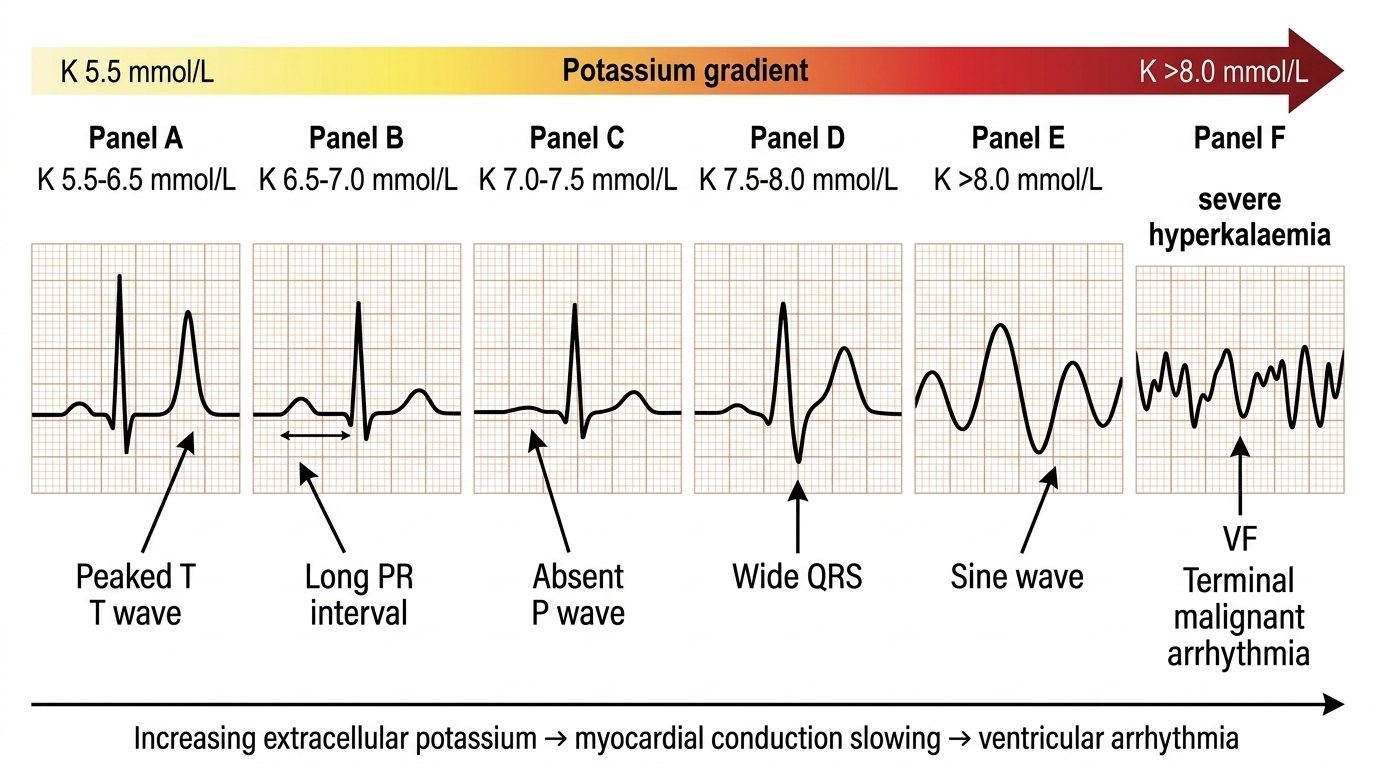
ECG Progression in Hyperkalaemia
SELF-CHECK
A 65-year-old man with CKD (eGFR 20) on lisinopril and spironolactone presents acutely unwell. Serum potassium is 7.2 mmol/L. ECG shows absent P waves and wide QRS complexes. His blood pressure is 90/60 mmHg. What is the FIRST immediate intervention?
A. Nebulised salbutamol 10 mg immediately
B. Intravenous insulin 10 units with 50% dextrose
C. Intravenous calcium gluconate 10 mL of 10% over 3–5 minutes
D. Urgent haemodialysis
Reveal Answer
Answer: C. Intravenous calcium gluconate 10 mL of 10% over 3–5 minutes
The ECG shows life-threatening hyperkalaemia with absent P waves and wide QRS — this is an imminent risk for ventricular fibrillation. The FIRST and most urgent step is membrane stabilisation with IV calcium gluconate. Calcium raises the depolarisation threshold of cardiac cells, protecting against arrhythmia. It acts within 1–3 minutes. Insulin/dextrose and salbutamol cause transcellular shift (acting in 15–30 minutes) and should follow immediately after calcium. Haemodialysis is definitive removal but takes time to set up. The sequence matters: stabilise first, then shift, then remove. Skipping membrane stabilisation risks cardiac arrest in the interval before insulin takes effect.
SELF-CHECK
A 55-year-old woman on long-term thiazide diuretic therapy for hypertension has a routine serum potassium of 2.8 mmol/L and a magnesium of 0.55 mmol/L (normal 0.70–1.05). She is given IV potassium replacement but her potassium remains at 2.9 mmol/L after 80 mmol replacement. What is the most likely explanation for refractory hypokalaemia?
A. Continued thiazide-driven renal K losses requiring higher replacement dose
B. Transcellular shift from concurrent alkalosis
C. Concurrent hypomagnesaemia causing renal K wasting
D. Pseudohypokalaemia from haemolysis during sampling
Reveal Answer
Answer: C. Concurrent hypomagnesaemia causing renal K wasting
Hypomagnesaemia is the classic cause of hypokalaemia refractory to potassium replacement. Magnesium is required to keep ROMK channels closed in the cortical collecting duct; when Mg²⁺ is depleted, ROMK channels are constitutively open, causing persistent renal K⁺ wasting. Potassium cannot be retained until magnesium is corrected. This combination — hypokalaemia + hypomagnesaemia — is classic in thiazide diuretic therapy and in alcoholism. Always measure and correct magnesium before concluding that potassium replacement is failing. IV magnesium sulphate (10–20 mmol over 30–60 minutes) typically restores the ability to retain K⁺.
CLINICAL PEARL
In hyponatraemia management, the danger lies not only in the sodium level but in the speed of change in either direction. A sodium that has fallen gradually over days allows the brain to adapt by extruding osmolytes; a sodium that has fallen acutely over hours (e.g., marathon runner drinking excessive water, postoperative hyponatraemia from SIADH) has not allowed adaptation — and acute symptomatic hyponatraemia requires urgent correction with hypertonic saline. But the chronic form requires caution: correct no faster than 10–12 mmol/L in 24 hours. If you have already corrected too fast, give 5% dextrose or desmopressin to relower the sodium and prevent osmotic demyelination syndrome. Stop — reassess — correct the overcorrection.
In hyperkalaemia, commit to the sequence: calcium first (membrane stabilisation) → insulin plus dextrose (shift) → dialysis or resins (removal). The ECG is your guide to urgency: peaked T waves alone require treatment but not immediate dialysis; absent P waves with wide QRS is a cardiac emergency requiring calcium within minutes. Never wait for a repeat potassium result before giving calcium to a patient with a dangerous ECG pattern.
Self-Assessment: Applying Sodium and Potassium Disorder Frameworks
The following scenarios integrate the diagnostic algorithms and management principles from this module. In each case, identify the mechanism, classify the disorder, and determine the correct management approach. Work through each scenario before reading the analysis — the objective is not merely to arrive at the correct answer but to follow the correct reasoning chain: serum osmolality first in hyponatraemia, then volume status, then urine chemistry; ECG first in hyperkalaemia before any blood result. The key discriminating skills tested here are: interpreting simultaneous serum and urine sodium and osmolality to classify hyponatraemia correctly; recognising when giving saline would worsen rather than help; sequencing hyperkalaemia interventions in the physiologically correct order; and applying the rate-of-correction rule in both sodium directions (too slow leaves the patient symptomatic; too fast causes osmotic demyelination or cerebral oedema). These are the skills that distinguish a physician who understands electrolyte physiology from one who has memorised values without framework. The scenarios below span the clinical spectrum from chronic ward-discovered hyponatraemia to acute ECG-change hyperkalaemia — both of which you will encounter in your first weeks as a house officer.
Provided image
Scenario A: A 45-year-old man with known heart failure (LVEF 25%) presents with increasing leg swelling and dyspnoea. Serum sodium is 128 mmol/L. Serum osmolality is 268 mOsm/kg. He has significant bilateral oedema and a raised JVP. Urine sodium is 8 mmol/L. What is the diagnosis and the correct immediate management?
Analysis: Hypervolaemic hypotonic hyponatraemia from heart failure — effective arterial hypovolaemia triggers non-osmotic ADH release and RAAS activation despite total body volume overload. Low urine Na confirms avid renal sodium retention. Management: fluid restriction (1000–1500 mL/day) + sodium restriction + optimise heart failure therapy (ACE inhibitor/ARB, beta-blocker, loop diuretic). Normal saline would worsen oedema. Hypertonic saline is contraindicated (the patient is already volume-overloaded).
Scenario B: A 30-year-old woman is admitted after a generalised tonic-clonic seizure. Serum sodium is 117 mmol/L. She had been drinking approximately 6 litres of water per day due to a psychiatric disorder. Urine osmolality is 45 mOsm/kg (very dilute). Urine sodium is 5 mmol/L. What is the diagnosis and the emergency management?
Analysis: Acute symptomatic hyponatraemia from psychogenic polydipsia (massive water intake overwhelms renal diluting capacity; urine is maximally dilute — low osmolality and low Na confirm kidneys are trying to excrete water). Treatment: 3% hypertonic saline 100 mL IV bolus — raise Na by 4–6 mmol/L acutely to terminate seizure activity. Then fluid restrict and allow spontaneous correction. Do NOT exceed 10–12 mmol/L in first 24 hours.
Scenario C: A 70-year-old man with type 2 diabetes, CKD (eGFR 18), and heart failure on lisinopril, frusemide, and spironolactone has a routine K⁺ of 6.3 mmol/L. ECG shows peaked T waves. He is otherwise asymptomatic. How do you manage?
Analysis: Moderate hyperkalaemia with ECG changes in a patient with multiple risk factors (CKD + RAAS blockade + K-sparing diuretic). Step 1: IV calcium gluconate (ECG changes present). Step 2: Insulin plus dextrose (shift K into cells). Step 3: Review and suspend potassium-elevating drugs (stop spironolactone, consider reducing/stopping ACE inhibitor after volume status assessment). Step 4: Loop diuretic if urine output adequate. If K⁺ does not fall or remains >6.0 with ongoing ECG changes, arrange urgent nephrology review regarding haemodialysis. Long-term: consider switching to a non-K-sparing diuretic.