Page 14 of 20
IM29.{24,26} | Impaired Physicians and Research Consent — SDL Guide (Part 2)
Vulnerable Populations, Illiterate Participants, and Special Consent Scenarios
The consent process described above applies to a competent, literate adult making an autonomous, fully informed decision. Real clinical research frequently involves participants who do not fit this straightforward profile — and the additional protections required in these circumstances are a key assessment point in the NMC IM29.26 competency. The ICMR Guidelines 2017/2023 and the Indian GCP specify detailed additional safeguards for vulnerable populations, defined as individuals who have reduced capacity for autonomous decision-making due to power imbalances, cognitive limitations, institutional dependency, or socio-economic circumstances.
The categories of vulnerability recognised in ICMR Guidelines with their specific additional requirements are:
Children (minors under 18 years): Consent is obtained from the parent or legal guardian (called permission in research ethics terminology). Assent — the child's own agreement to participate, in age-appropriate terms — must also be sought from any child aged 7 years or older who is capable of understanding, and from all adolescents. A child's dissent (explicit refusal) overrides parental permission in research not offering direct benefit. The distinction is clinically important: parental permission alone is insufficient for research involving minors who are capable of expressing a view.
Illiterate adults: As discussed in the companion SDL on medical ethics, illiteracy does not remove the right to research participation — it requires a procedurally modified consent. The ICMR-specified procedure for an illiterate adult participant: (a) the Participant Information Sheet must be read aloud in the participant's language, with adequate explanation; (b) an impartial witness — independent of the research team and the participant's family — must be present throughout the consent process; (c) the participant's signature is replaced by their left thumb impression (or other identifiable mark); (d) the impartial witness signs the consent form, attesting that the information was given and the participant appeared to understand and consent voluntarily; (e) the consent form must clearly state that the participant is illiterate and the witness name and relationship must be documented.
Patients enrolled in research by their treating clinician: The therapeutic relationship creates a power differential — patients may feel unable to refuse participation when their doctor asks them to enrol. The ICMR Guidelines require that the consent process explicitly address this: the participant must be clearly told that declining to participate will not in any way affect the quality or availability of their clinical care, and this assurance should ideally be provided by someone other than the treating clinician.
Psychiatric patients: Capacity assessment is mandatory before enrolling any patient with a psychiatric diagnosis into research. An acute psychiatric episode (psychosis, severe depression) may temporarily remove the ability to provide valid consent. Research on patients who are compulsorily admitted under the MHCA 2017 is permitted only with approval from the treating team, the participant's nominated representative, and the IEC — and only for research on the mental health condition itself, not for unrelated studies.
Prisoners and institutional residents: Participation must be genuinely voluntary — coercion from the institutional authority or offers of benefits that constitute undue inducement (e.g., parole consideration) are prohibited. Research in these settings requires specific IEC review and approval.
Emergency research (when consent cannot be obtained): Research involving emergency care patients (e.g., a trial of a novel CPR intervention in cardiac arrest) may proceed without prospective consent under a waiver of consent approved by the IEC, provided: (a) the research could not be conducted in non-emergency populations; (b) the intervention poses minimal additional risk beyond standard emergency care; (c) consent will be obtained retrospectively from the participant or their legally authorised representative as soon as possible; and (d) the IEC has specifically approved the waiver.
The practical simulation of research consent (IM29.26) — the component tested in the OSCE — requires the student to demonstrate the full ten-step consent process described above, adapted to the specific participant scenario presented. Key performance indicators that OSCE assessors evaluate:
| OSCE criterion | Performance standard |
|---|---|
| Introduces self and role | Name, designation, and affiliation with study team stated clearly |
| Explains research vs treatment distinction | Explicitly corrects therapeutic misconception if present |
| Covers all 10 ICMR elements | Each element addressed in plain language |
| Assesses comprehension | Asks participant to re-explain in own words (not just 'Do you understand?') |
| Confirms voluntariness | Explicitly states right to refuse and right to withdraw |
| Handles illiterate participant correctly | Thumb impression + impartial witness procedure demonstrated |
| Responds to a refusal | Accepts refusal graciously; confirms no impact on clinical care |
| Documentation | Consent form elements correctly completed with date/time/witness |
SELF-CHECK
You are the sub-investigator in a Phase II trial of a new antihypertensive drug. A potential participant, a 70-year-old retired teacher who is fully literate, has read the participant information sheet and says: 'I don't want to be on a placebo — I want to be sure I get the real drug.' He is currently uncontrolled on two antihypertensives and genuinely needs better blood pressure control. He says he will only sign the consent if you guarantee he will receive the active drug. How do you respond?
A. Reassure him that he will receive the active drug to obtain his consent — the trial needs participants
B. Explain clearly that the study is double-blind and randomised, that neither you nor he can know his allocation, that standard care will continue regardless of allocation, and that he should only participate if he understands and accepts the randomisation
C. Exclude him from the study — his conditional consent is invalid and he should not be enrolled
D. Obtain his conditional consent and note in the file that he wishes to receive the active drug — the randomisation will override his preference anyway
Reveal Answer
Answer: B. Explain clearly that the study is double-blind and randomised, that neither you nor he can know his allocation, that standard care will continue regardless of allocation, and that he should only participate if he understands and accepts the randomisation
This is the therapeutic misconception scenario — the participant wants to use research enrolment as a pathway to better treatment. The correct response is complete transparency: explain clearly that the study is double-blind randomised controlled, that you cannot guarantee his allocation, and that his standard clinical care (optimisation of his current antihypertensives) will continue irrespective of his trial participation. If after this explanation he still only wishes to consent conditionally on receiving the active drug, his consent is not valid — he cannot be enrolled with the understanding that his preference overrides randomisation. Options A and D involve deception or documentation of invalid consent, which are serious GCP violations. Option C is premature — the patient should be given the correct information first and given the opportunity to consent on a fully informed basis; only if he still refuses to accept randomisation should he be excluded.
SELF-CHECK
A Phase III trial is being conducted on a new antimalarial drug. An 8-year-old child is identified as a potential participant. The child's father signs the consent form and says the child is 'fine with it.' The child, when asked directly, says 'I don't want to take any more tablets — the last ones made me feel sick.' According to ICMR Guidelines and research ethics principles, what should happen?
A. Proceed with enrolment — parental consent overrides child's expressed preference in all cases
B. Ask the father to persuade the child — parental authority is determinative
C. The child's dissent should be respected; she should not be enrolled in research that does not offer her direct benefit unless the IEC has specifically waived the assent requirement
D. Enrol the child since her concern is about side effects from a previous drug, not about this trial
Reveal Answer
Answer: C. The child's dissent should be respected; she should not be enrolled in research that does not offer her direct benefit unless the IEC has specifically waived the assent requirement
ICMR Guidelines and the Declaration of Helsinki (2013) require that children aged 7 or older who are capable of understanding be asked for assent, and that a child's dissent — an explicit refusal — should be respected in research that does not offer the child direct benefit. An 8-year-old expressing clear reluctance ('I don't want to') is a dissent that must be taken seriously. The fact that her concern may relate to a previous drug's side effects is relevant context to explore, but does not override her right to refuse participation. If the trial is of direct benefit to the child (e.g., she is being treated for malaria and the trial drug is the only realistic treatment option), the ethical calculus may differ — but in a general Phase III trial of a new antimalarial where her participation is for population benefit rather than her personal treatment, her dissent is ethically decisive. Parental permission is necessary but not sufficient when the child has expressed a clear dissent.
Self-Assessment: Integrating Impaired Physician Response and Research Consent
You have now covered the full scope of IM29.24 and IM29.26 — the definition and causes of physician impairment, the ethical and legal frameworks governing the response to an impaired colleague, the regulatory foundation and procedural elements of research consent, and the specific adaptations required for vulnerable populations including illiterate adults and children. The self-assessment below consolidates this through three integrative scenarios that combine both topics and require you to navigate the professional obligations, the relevant regulatory framework, and the practical communication skills.
For each scenario, identify the applicable regulatory framework (NMC Act 2020, MHCA 2017, ICMR Guidelines 2017/2023, GCP, Declaration of Helsinki, or a combination), state the ethical principles engaged, and describe the specific action you would take.
Scenario A — The impaired colleague with mental health condition:
Dr. Sunita, a third-year psychiatry resident, discloses to you in confidence that she has been diagnosed with major depressive disorder for the past six months, is on antidepressants, and is seeing a psychiatrist fortnightly. She is managing her workload and her department head is unaware. She asks you not to tell anyone. She is not showing any signs of clinical impairment. What are your ethical and legal obligations?
Analysis: This scenario tests the intersection of IM29.24 and MHCA 2017. The MHCA 2017 prohibits discrimination on grounds of mental health condition and protects individuals' right to seek and receive mental health treatment without mandatory reporting to their employer. More importantly, IM29.24 is triggered by impairment affecting patient care — not by diagnosis. Dr. Sunita is managing her condition, is under appropriate professional care, and is showing no signs of clinical impairment. Your obligations here are: (a) maintain the confidentiality of her disclosure under the professional duty of confidentiality (NMC Code); (b) continue to observe her clinical function — if you subsequently observe signs of impairment, the situation changes; (c) support her emotionally and professionally; (d) do not report to her department head — there is currently no patient safety risk to trigger that obligation. If she were showing signs of impairment, the calculus would change — but depression with treatment and a functioning clinician is not the same as an impaired clinician.
Scenario B — The research consent in a challenging setting:
You are sub-investigator in a study of a new SGLT2 inhibitor for type 2 diabetes with CKD. A potential participant is a 55-year-old woman with limited formal education. Her husband is present and has read the participant information sheet on her behalf. He says she agrees. She has not been spoken to directly. You notice she appears to understand the conversation around her but has not spoken. How do you proceed?
Analysis: This is the proxy-consent-before-patient-speaks scenario — a form of surrogate consent being applied to a potentially competent adult. The correct approach: speak to the participant directly, in her language, using the participant information sheet. Capacity assessment first — she may be fully competent and merely deferential. If she has capacity, her own consent is required; her husband's reading of the PIS does not substitute for direct consent. Explain each ICMR element directly to her, ask her comprehension questions, and ensure her consent is genuinely voluntary — not driven by pressure from her husband or the treating team. If she lacks capacity (confirmed by assessment, not assumed from her silence), the surrogate consent process applies with her husband as nearest relative, and her assent should still be sought for any aspect she can understand.
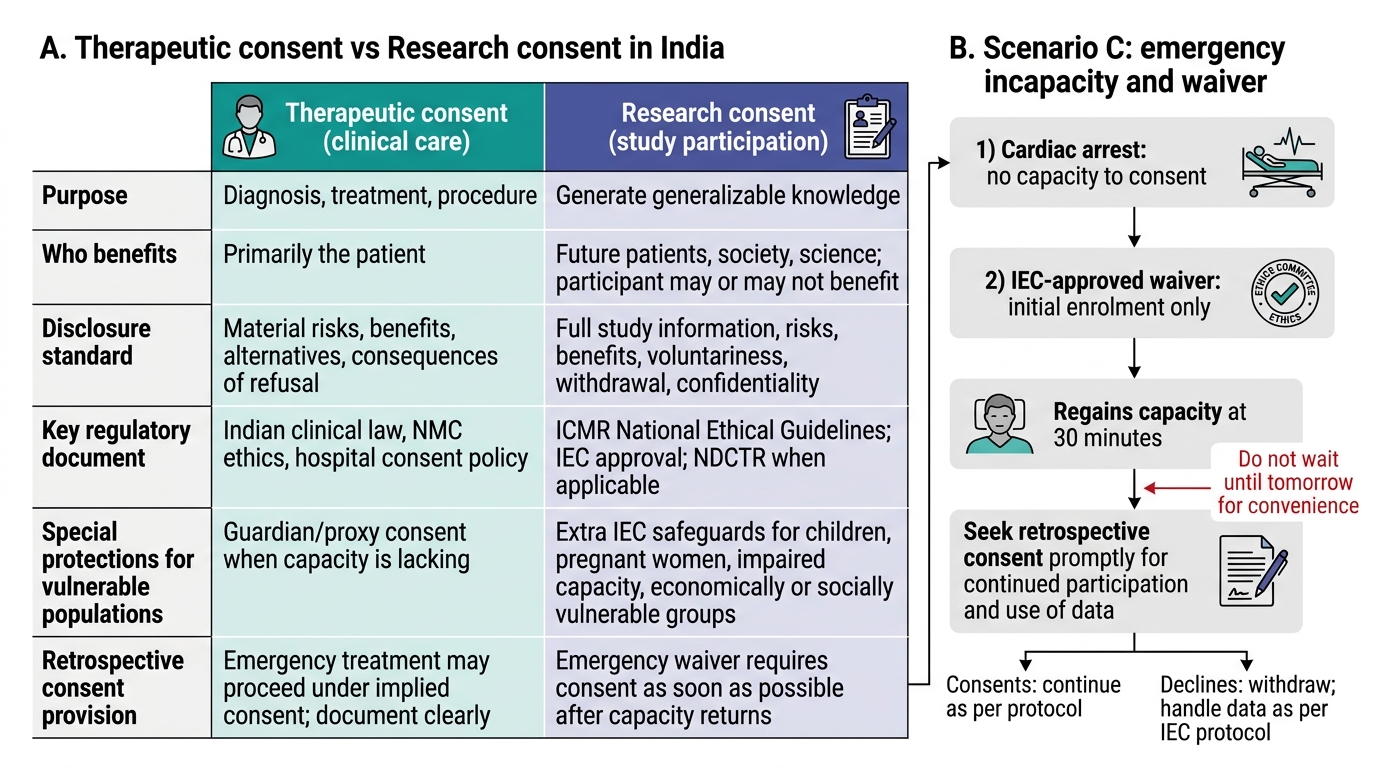
Therapeutic Consent vs Research Consent in India
Scenario C — Emergency incapacity and waiver of consent in research:
A cardiac arrest study is being conducted in your hospital's emergency department. The protocol has IEC-approved waiver of consent for the initial resuscitation phase (the patient cannot consent during active cardiac arrest). A patient is enrolled under the waiver. She survives and regains capacity 30 minutes after arrival. The research assistant tells you she will be told about the study 'when she feels better — maybe tomorrow.' What is the correct procedure?
Analysis: The IEC-approved waiver of consent for emergency research requires retrospective consent to be obtained 'as soon as possible' — not at clinical convenience. ICMR Guidelines specify that when the participant regains capacity, consent must be sought promptly for continued participation in the study and for use of any data collected during the waiver period. If the patient declines consent retrospectively, the study data from her participation may not be usable (depending on the IEC protocol), and she must be withdrawn from the study immediately. 'Tomorrow when she feels better' is a procedural delay that could be challenged as prolonging non-consented research participation. The correct action: the principal investigator or sub-investigator should visit the patient within the next 1–2 hours (once she is clinically stable and capable), explain the study and the waiver procedure, and obtain prospective consent for continued participation or withdrawal.
CLINICAL PEARL
On impaired physicians: the most dangerous impaired colleague is not the one who is obviously intoxicated — it is the one who is functioning just well enough that peers rationalise not acting. The clinical culture that says 'he's under a lot of stress — we should give him space' rather than 'his decisions are inconsistent and patients may be at risk — we need to act' is a culture in which preventable harm is normalised. Acting on a reasonable suspicion of impairment is not a betrayal of a colleague; it is the most protective thing you can do for them and for their patients. An early intervention — before a serious adverse event occurs — is far better than a NMC fitness to practise hearing after one does.
On research consent: the phrase 'therapeutic misconception' describes one of the most common errors in research consent — the participant's belief that they are receiving the best treatment available, rather than participating in an experiment. A single direct question at the beginning of the consent conversation — 'Do you understand that this is a research study, and that the treatment you may receive might be a placebo or a new drug that may not work?' — often reveals whether therapeutic misconception is present. Correcting it is the foundation of valid research consent.