Page 6 of 18
MI4.5-6 | Food Poisoning & Acid-Peptic Disease (H. pylori) — SDL Guide (Part 2)
Helicobacter pylori — Aetiology & Pathogenesis of APD
Helicobacter pylori Pathogenesis in Acid Peptic Disease
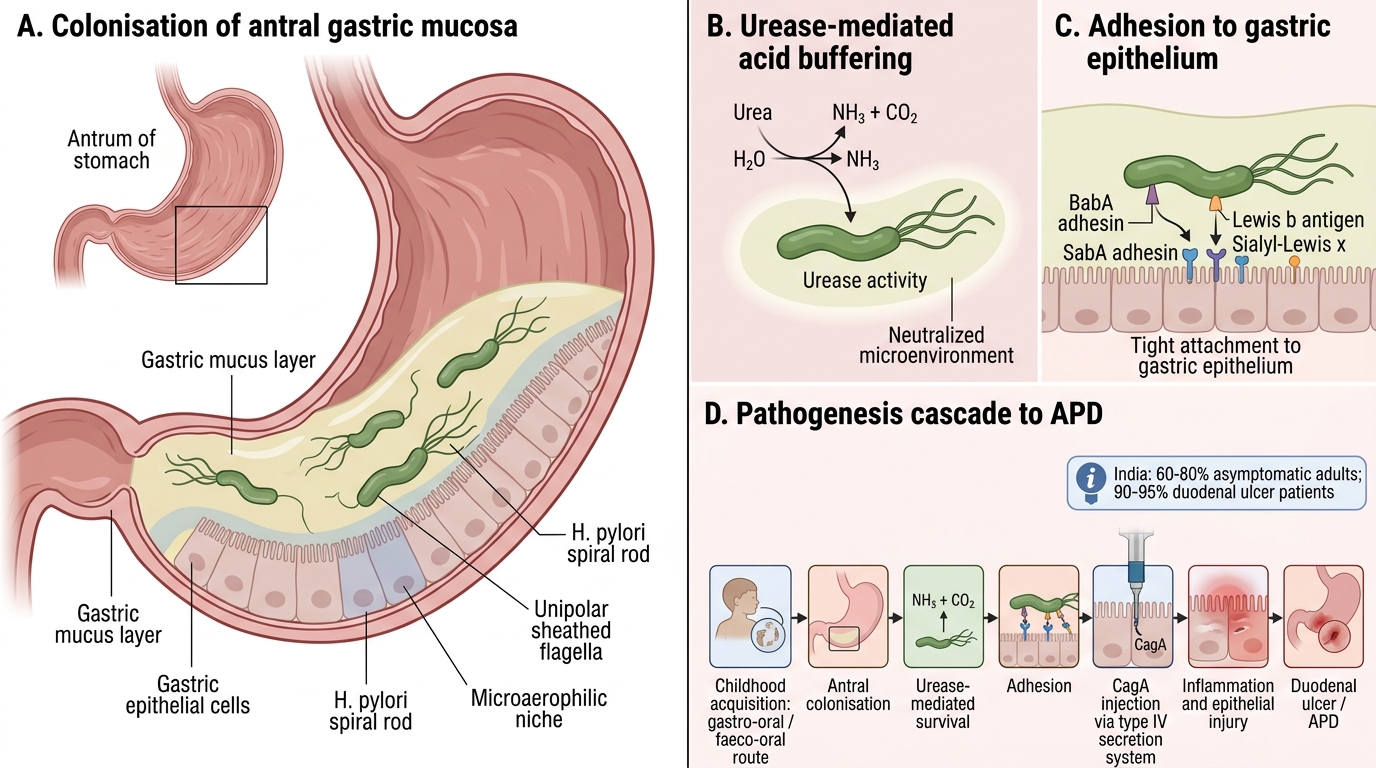
Helicobacter pylori is a Gram-negative, microaerophilic, spiral/curved rod (helix-shaped), 3–5 µm long, with multiple unipolar sheathed flagella and a characteristic urease activity so powerful it is the basis of nearly every diagnostic test.
Epidemiology: Prevalence in India — ~60–80% of asymptomatic adults; ~90–95% of duodenal ulcer patients. Acquisition typically in childhood via gastro-oral or faeco-oral route. Higher in lower socio-economic groups (shared water, crowded households).
Pathogenesis cascade:
1. Colonisation of antral gastric mucosa: urease splits urea → NH₃ → buffers the acid environment, enabling survival
2. Adhesins (BabA, SabA) bind Lewis b blood group antigen and sialyl-Lewis x on epithelium
3. CagA (cytotoxin-associated gene A) — injected via type IV secretion system → disrupts tight junctions, activates oncogenic signalling
4. VacA (vacuolating cytotoxin) — forms pores in epithelial cells → vacuolation → cell death
5. Neutrophil recruitment → IL-8 release → mucosal inflammation → chronic active gastritis
6. Increased acid secretion (↑gastrin from inflamed antrum, ↓somatostatin) → duodenal ulcer
7. Atrophic gastritis over decades → intestinal metaplasia → gastric cancer (IARC Group 1 carcinogen)

Pathogenesis of Helicobacter pylori Infection
Laboratory Diagnosis of H. pylori
⚑ AI image — pending faculty review (auto-QA score 7/10; best of 3 attempts)
Laboratory Diagnosis of Helicobacter pylori
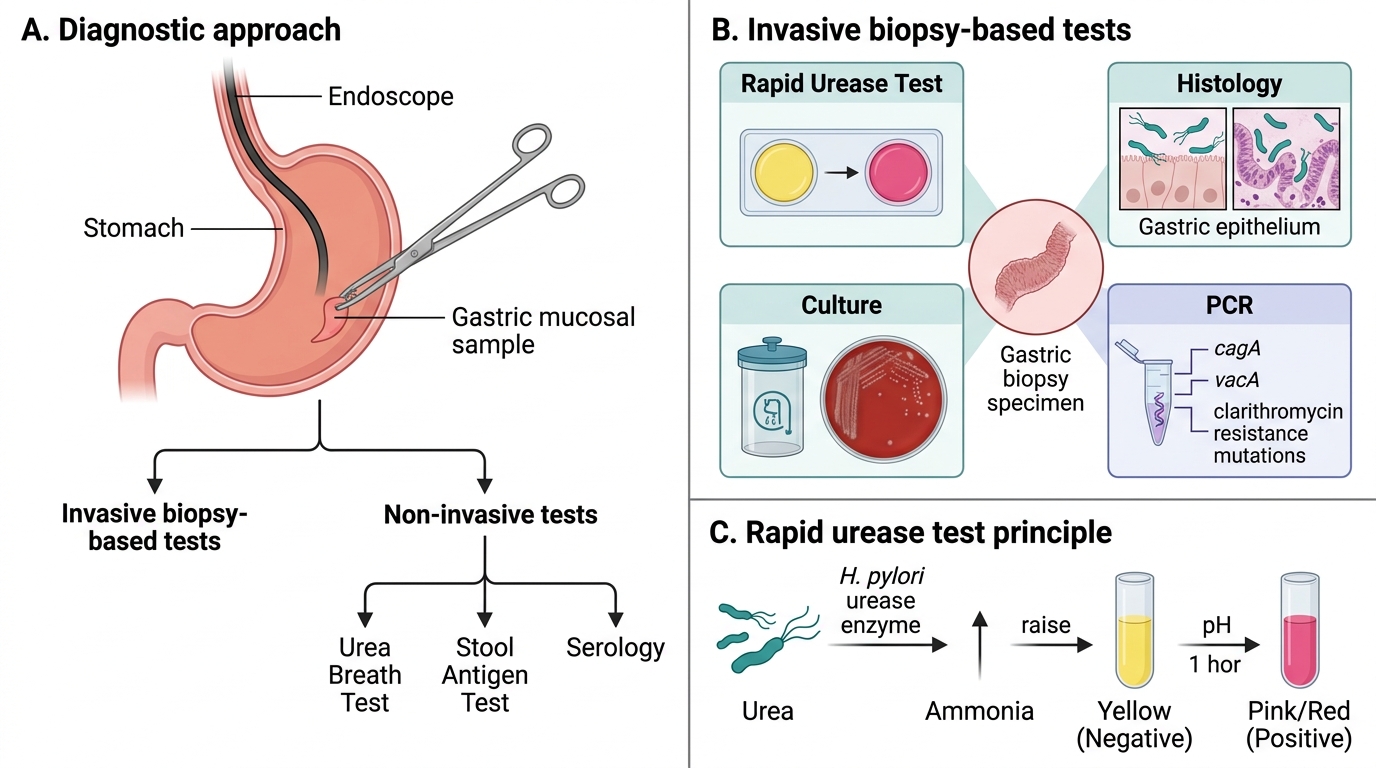
Diagnostic tests divide into invasive (require endoscopy + biopsy) and non-invasive:
Invasive (biopsy-based):
| Test | Principle | Notes |
|---|---|---|
| Rapid Urease Test (CLO test) | Urease splits urea → NH₃ → pH ↑ → colour change (yellow → pink/red) within 1 h | Most widely used; 90% sensitivity |
| Histology (Giemsa/Warthin-Starry) | Visualise curved rods on gastric epithelium | Gold standard for tissue pathology |
| Culture | Microaerophilic (5–10% CO₂), 37°C, 3–7 days; Columbia blood agar | Reference; antibiotic sensitivity testing |
| PCR | Detects cagA, vacA; clarithromycin resistance mutations | Increasing use in refractory cases |
Non-invasive:
| Test | Principle | Notes |
|---|---|---|
| Urea Breath Test (UBT) | Oral ¹³C- or ¹⁴C-urea → urease → labelled CO₂ in breath | Best for post-treatment eradication check; 95% accuracy |
| Stool Antigen Test (HpSA) | EIA detects H. pylori antigen in stool | Accuracy comparable to UBT; stop PPIs 2 weeks prior |
| Serology (IgG ELISA) | Detects anti-H. pylori IgG | Cannot distinguish active vs. past infection; NOT for eradication check |
Management: Triple therapy — PPI + clarithromycin + amoxicillin × 14 days (first-line in India where clarithromycin resistance <20%); quadruple therapy for clarithromycin-resistant strains.
SELF-CHECK
A 50-year-old man was successfully treated for H. pylori 4 weeks ago with triple therapy. The physician wants to confirm eradication. Which test is MOST appropriate at this stage?
A. IgG ELISA serology for H. pylori
B. Urea breath test (UBT) after stopping PPI for 2 weeks
C. Rapid urease test on antral biopsy
D. Serum gastrin level
Reveal Answer
Answer: B. Urea breath test (UBT) after stopping PPI for 2 weeks
The urea breath test (UBT) is the preferred non-invasive test for confirming H. pylori eradication as it detects active urease activity from live organisms. It should be performed at least 4 weeks after completing antibiotics and 2 weeks after stopping PPIs (which can suppress urease activity and cause false negatives). Serology (IgG ELISA) remains positive for months to years after eradication and is useless for post-treatment confirmation. Repeat endoscopy with rapid urease test is invasive and not first-line unless symptoms recur.
CLINICAL PEARL
The triple-threat of CagA: Not all H. pylori strains are equally virulent. Strains carrying the cagA gene (the 'pathogenicity island') cause more severe gastritis, higher ulcer rates, and a significantly elevated risk of gastric adenocarcinoma. CagA is injected directly into epithelial cells, where it activates the oncogenic Ras–ERK signalling pathway. CagA-positive strains predominate in East and South Asia—explaining the paradox of high H. pylori prevalence yet higher gastric cancer rates in Asian populations despite lower rates of smoking.