Page 5 of 22
OG23.2 | Delayed Puberty — SDL Guide
Learning Objectives
- Define delayed puberty in girls using the standard age thresholds (no B2 by 13 yr; no menarche by 15 yr)
- Classify delayed puberty into hypogonadotropic hypogonadism, hypergonadotropic hypogonadism, and eugonadism with outflow obstruction
- Enumerate the common causes of each category with their distinguishing clinical features
- Describe a structured investigation pathway for a girl presenting with delayed puberty
- Outline the management principles for the most common causes: CDGP, Turner syndrome, Kallmann syndrome, and outflow tract obstruction
INSTRUCTIONS
Delayed puberty is one of the most clinically important presentations in adolescent gynaecology. It encompasses conditions ranging from benign constitutional delay — requiring only reassurance — to Turner syndrome, gonadal failure requiring lifelong hormone replacement, and anatomical obstruction requiring surgery. Getting the workup right, efficiently and without unnecessary anxiety for the patient and family, depends on a logical classification-driven approach. This module equips you with the framework to classify, investigate, and manage the full spectrum of delayed puberty.
References
- DC Dutta's Textbook of Gynecology, 8th ed., Ch. 26 — Disorders of Puberty (textbook)
- Shaw's Textbook of Gynaecology, 17th ed., Ch. 7 — Puberty and Its Disorders (textbook)
- Williams Gynecology, 4th ed., Ch. 14 — Pediatric and Adolescent Gynecology (textbook)
- FOGSI Guidelines on Evaluation of Delayed Puberty in Adolescent Girls (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 15-year-old girl is brought by her mother who is worried she has 'not developed like her friends.' On examination you find no breast development (Tanner B1), no pubic hair, and a height at the 3rd centile. She has a webbed neck, a wide carrying angle, and a shield-shaped chest. FSH comes back at 68 IU/L. What is the diagnosis, and what does this patient need for the rest of her life?
WHY THIS MATTERS
Delayed puberty causes significant psychological distress to adolescent girls and their families. However, the consequences go far beyond the emotional: untreated gonadal failure leads to osteoporosis and fracture risk from oestrogen deficiency; Turner syndrome carries a 30–50 times increased risk of aortic dissection and requires cardiac surveillance; Kallmann syndrome is associated with infertility without specialist intervention. Constitutional delay — the most common cause — requires nothing more than reassurance, but only after pathological causes are confidently excluded. This module gives you the classification framework and investigation pathway to confidently separate the benign from the serious.
RECALL
Recall from the previous module (og12-puberty): delayed puberty is defined as no breast development (B2) by age 13, or no menarche by age 15, or more than 5 years from thelarche to menarche. The HPO axis can fail at three levels — hypothalamic/pituitary (low FSH/LH), gonadal (high FSH/LH, the pituitary signals but receives no response), or there can be normal hormones with anatomical obstruction. FSH and LH measurement is therefore the pivotal first investigation.
Clinical Presentation of Delayed Puberty
Delayed puberty in girls is defined by the failure to reach expected pubertal milestones within the normal age range. The accepted clinical thresholds are: (1) no breast development (Tanner B2) by age 13 years, (2) no menarche by age 15 years in a girl who has already developed secondary sex characteristics, or (3) more than 5 years elapsing between the onset of breast development and menarche. These thresholds represent approximately 2–2.5 standard deviations below the population mean and define the point at which investigation is warranted rather than watchful waiting.
The clinical presentation is most commonly that of a concerned parent or the girl herself presenting because peers have 'developed and she has not.' The history should establish the exact chronology of any pubertal changes that have occurred, the girl's growth pattern over the preceding years, family history (especially the father's pubertal timing — CDGP is familial), anosmia (Kallmann syndrome), cyclical abdominal pain (outflow obstruction), chronic illness, nutritional history, and exercise intensity. These elements are not merely context — each one points toward or away from a specific diagnostic category.
On examination, the priority findings are: (a) Tanner staging of breast and pubic hair — are secondary sex characteristics absent (hypergonadotropic failure) or present with absent menarche (outflow obstruction or late hypogonadotropic delay)? (b) Height and weight — short stature with delayed puberty strongly suggests Turner syndrome; (c) Somatic stigmata of Turner syndrome — webbed neck, wide carrying angle (cubitus valgus), shield chest, widely spaced nipples, low posterior hairline, lymphoedema; (d) Anosmia — tested by asking the patient to identify a familiar smell with eyes closed (Kallmann syndrome); (e) Pelvic examination — presence of the uterus and vagina (rules out MRKH and androgen insensitivity).
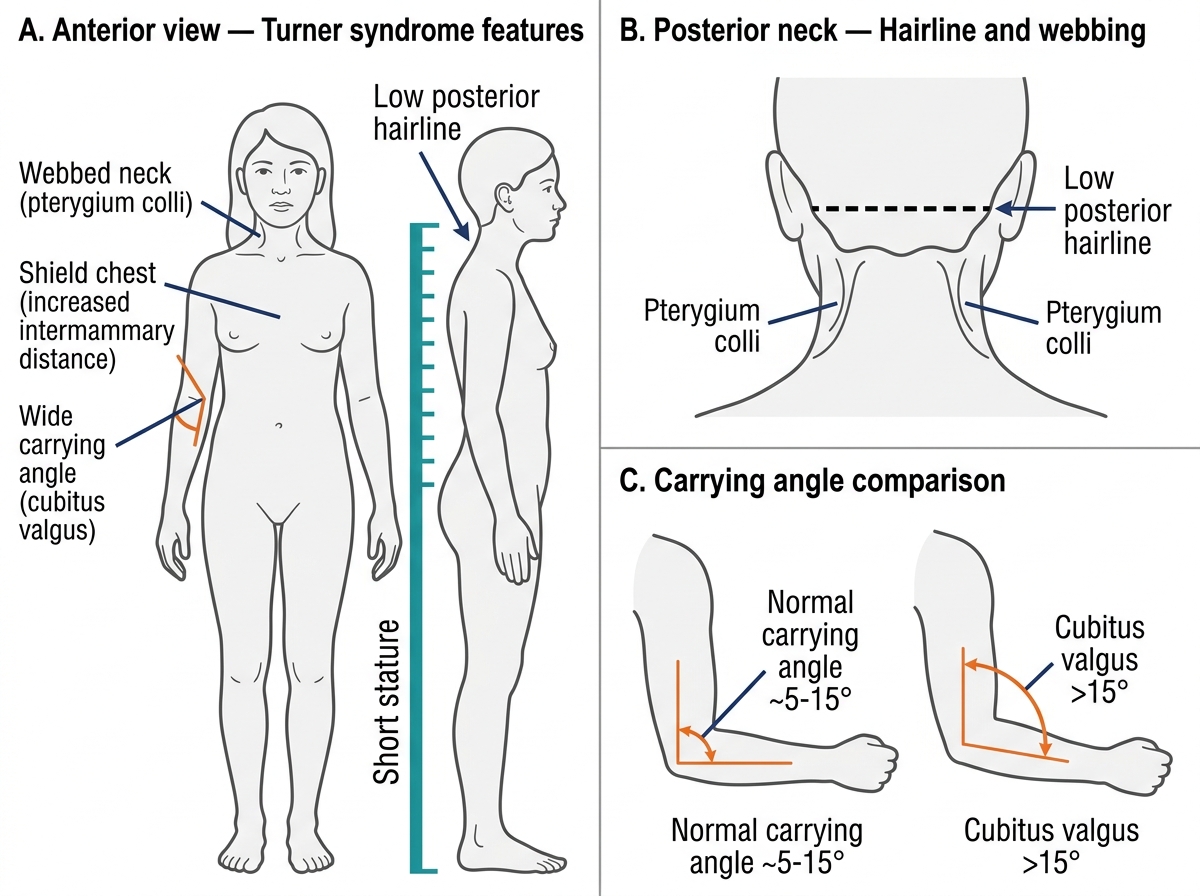
Somatic Features of Turner Syndrome (45,X)
Pathophysiology and Classification of Delayed Puberty
The classification of delayed puberty is anchored to the FSH and LH measurement, which immediately localises the level of failure within the HPO axis. This is the single most important diagnostic step because it divides delayed puberty into three mechanistically distinct categories with entirely different management pathways. Understanding the mechanism behind each category transforms a potentially confusing list of eponymous conditions into a logical, predictable framework. Once you grasp that the HPO axis can fail at the hypothalamic-pituitary level (no signal sent to the ovary), the gonadal level (signal sent but ovary cannot respond), or the anatomical outflow level (ovary responds but blood cannot exit), every cause of delayed puberty falls naturally into place.
The FSH/LH result should always be interpreted alongside the clinical picture — particularly the presence or absence of secondary sex characteristics. A girl with no secondary sex characteristics at all has had no oestrogen production at any point, pointing to either very early-onset gonadal failure or central failure from childhood. A girl with normal breast and pubic hair development but absent menstruation has had normal oestrogen production, narrowing the differential to outflow obstruction or a very late-onset central cause. This clinical correlation prevents the error of over-relying on a single laboratory value without integrating the physical examination findings.
Category 1 — Hypogonadotropic Hypogonadism (low FSH and LH): The hypothalamic-pituitary unit is not activating the HPO axis. The ovaries are structurally normal but are not receiving gonadotrophin stimulation.
- Constitutional Delay of Growth and Puberty (CDGP): The most common cause of delayed puberty overall (~60–65% of all delayed puberty). A benign, familial variant — the HPO axis activates spontaneously but later than average. Bone age is delayed (typically 2–3 yr below chronological age). Positive family history in ~50–75% (father often had delayed puberty). FSH and LH are low-normal for chronological age but appropriate for bone age. Resolves entirely without treatment; eventual adult height is normal. Requires exclusion of Kallmann syndrome (anosmia absent in CDGP) and pituitary lesions.
- Kallmann syndrome: GnRH deficiency due to failure of GnRH neuron migration from the olfactory placode to the hypothalamus. Hallmark: hypogonadotropic hypogonadism + anosmia (absent sense of smell — X-linked most common form, KAL1 gene). Puberty does not occur spontaneously. MRI may show absent olfactory bulbs. Treatment requires GnRH pulsatile therapy or gonadotrophin replacement for fertility.
- Functional hypothalamic suppression: Undernutrition (anorexia nervosa — BMI <17.5), excessive exercise (female athlete triad: low energy availability + amenorrhoea + low bone density), chronic systemic illness. The GnRH pulse generator is suppressed by low leptin and elevated cortisol. Reversible with weight restoration and reduced exercise load.
- Pituitary lesions: Craniopharyngioma, prolactinoma (elevated prolactin suppresses GnRH), empty sella, post-irradiation hypopituitarism.
Category 2 — Hypergonadotropic Hypogonadism (high FSH and LH): The pituitary is generating gonadotrophin signals but the ovaries cannot respond — gonadal failure. FSH is markedly elevated (often >40 IU/L), LH is also elevated.
- Turner syndrome (45,X): The most common cause of primary amenorrhoea and hypergonadotropic hypogonadism. Streak ovaries (fibrous remnants devoid of follicles) produce no oestrogen; FSH and LH are very high. Classic somatic features: short stature, webbed neck, shield chest, wide carrying angle, widely-spaced nipples, lymphoedema. Cardiovascular: bicuspid aortic valve (~30%), aortic coarctation (~10%), risk of aortic root dilation — cardiac MRI essential. Karyotype confirms: 45,X in ~50%; mosaic (45,X/46,XX) in ~30–40% (milder phenotype).
- Premature Ovarian Insufficiency (POI): Ovarian follicle pool depleted or dysfunctional before age 40 (primary before menarche in delayed puberty context). Causes: autoimmune (anti-ovarian antibodies, associated with autoimmune thyroiditis), iatrogenic (chemotherapy — especially alkylating agents; pelvic irradiation), fragile X premutation (FMR1), galactosaemia.
- Swyer syndrome (46,XY gonadal dysgenesis): Phenotypic female with 46,XY karyotype; streak gonads; absent testes; primary amenorrhoea. Important: streak gonads with Y chromosome carry a 25–35% risk of gonadoblastoma — prophylactic gonadectomy is mandatory. External phenotype female because no testosterone/AMH was produced in utero.
Category 3 — Eugonadism with Outflow Tract Obstruction (normal FSH and LH): Ovarian function is normal, oestrogen is produced, secondary sex characteristics develop, but menstrual blood cannot exit.
- Imperforate hymen: Congenital membrane with no opening; haematocolpos forms. Presents with primary amenorrhoea + cyclical pelvic pain + bluish bulging hymen. FSH/LH normal; oestradiol normal.
- MRKH syndrome (Mayer-Rokitansky-Küster-Hauser): Congenital absence of the uterus and upper two-thirds of the vagina; 46,XX karyotype; normal ovaries and normal oestrogen. Presents as primary amenorrhoea with normal breast and pubic hair. Associated renal anomalies in ~30% (horseshoe kidney, unilateral renal agenesis — imaging essential).
- Androgen Insensitivity Syndrome (AIS): 46,XY; normal testes (produce testosterone and AMH); end-organ resistance to androgen. Phenotypically female. Complete AIS: absent uterus, absent/sparse pubic hair, blind-ending vagina; testes may be inguinal. FSH normal, testosterone male-range. Gonadectomy after puberty (oestrogen produced by peripheral conversion; delay until after breast development).
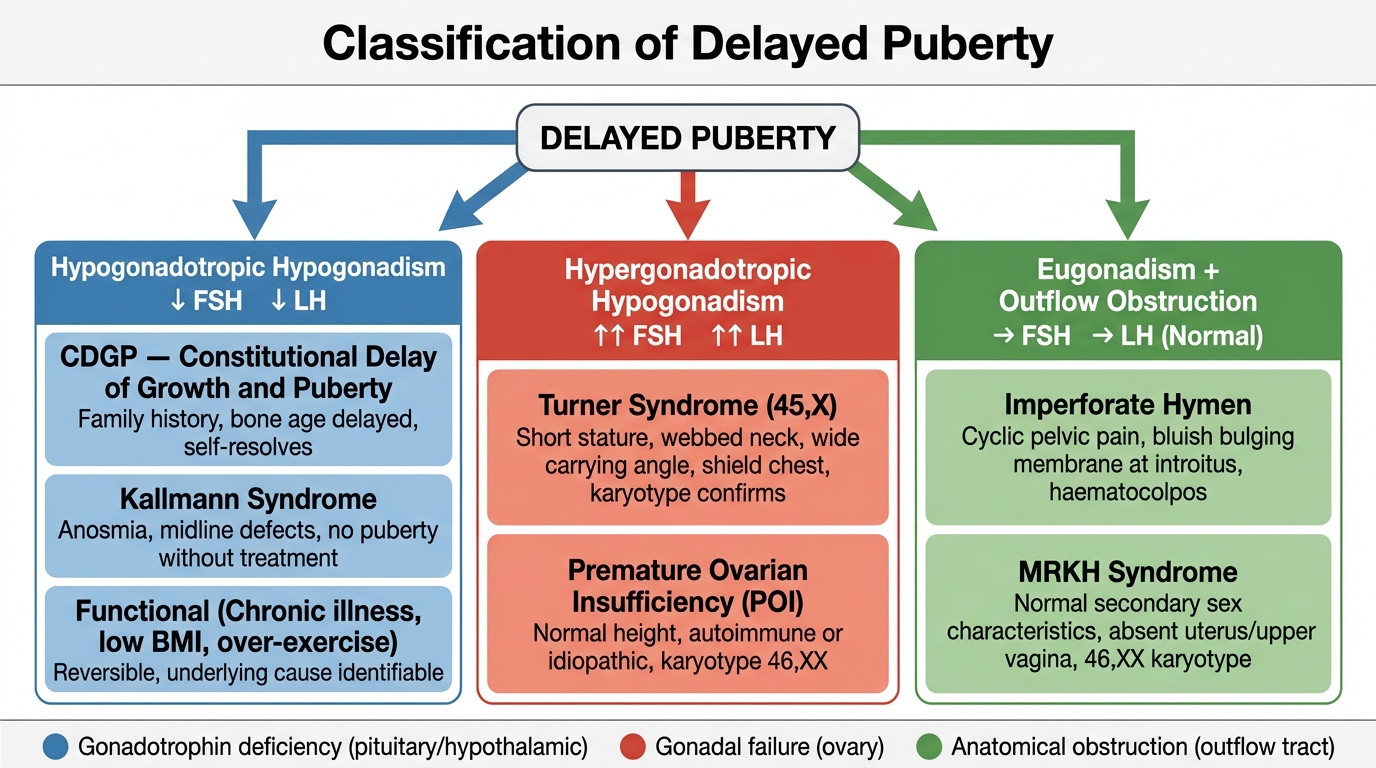
Classification of Delayed Puberty by Gonadotrophin Level
SELF-CHECK
A 14-year-old girl presents with primary amenorrhoea and no breast development. FSH is 75 IU/L, LH is 38 IU/L. She is 145 cm tall with a webbed neck and wide carrying angle. The most likely diagnosis is:
A. Constitutional delay of growth and puberty (CDGP)
B. Kallmann syndrome
C. Turner syndrome (45,X)
D. MRKH syndrome
Reveal Answer
Answer: C. Turner syndrome (45,X)
Very high FSH and LH indicate hypergonadotropic hypogonadism (gonadal failure). Combined with short stature and Turner somatic stigmata (webbed neck, wide carrying angle), the diagnosis is Turner syndrome. CDGP and Kallmann syndrome both have LOW gonadotrophins. MRKH has NORMAL FSH/LH and normal secondary sex characteristics.
Diagnosis and Investigation of Delayed Puberty
The investigation of delayed puberty follows a structured stepwise pathway directed by the clinical findings and the FSH/LH result. The goal is efficient classification into one of the three categories, followed by targeted confirmatory tests. Unnecessary investigations waste time and increase patient anxiety, but missing key tests — particularly karyotype in a short girl with high FSH — can delay a life-altering diagnosis. Every investigation ordered should be justified by the clinical question it answers, and the sequence matters: FSH/LH is cheap, fast, and immediately bifurcates the management pathway, so it must always be the first step. Specialist investigations (karyotype, MRI, GnRH stimulation test) come after this initial result directs the pathway.
It is worth noting that investigation and management are not always strictly sequential in practice. A girl with clear somatic features of Turner syndrome should have a karyotype ordered simultaneously with FSH/LH — there is no clinical logic in waiting for the FSH result when the diagnosis is already strongly suspected on clinical grounds. Similarly, a girl with cyclical pain and a bluish bulging hymen should be referred for surgical assessment without waiting for hormone results. The investigation framework below represents the default pathway for a presentation where the diagnosis is not immediately clinically apparent.
The investigation strategy proceeds as follows:
Step 1 — Initial blood tests (all cases):
- FSH and LH (the pivotal bifurcation test)
- Oestradiol (very low in both hypo- and hypergonadotropic failure)
- Prolactin (elevated in prolactinoma — a reversible cause of hypogonadotropic delay)
- Thyroid function — hypothyroidism can delay puberty
- FBC, ESR, renal and liver function — screen for chronic illness
- Bone age X-ray (left wrist and hand) — delayed bone age supports CDGP; advanced bone age with early puberty suggests precocious puberty
Step 2 — If FSH/LH are HIGH (hypergonadotropic):
- Karyotype — mandatory: Turner 45,X; mosaic Turner 45,X/46,XX; Swyer 46,XY (gonadectomy if Y material found)
- Anti-ovarian antibodies, anti-adrenal antibodies (autoimmune POI)
- FMR1 premutation testing (fragile X-associated POI)
- Pelvic ultrasound (streak ovaries in Turner; uterine presence)
- Cardiac MRI or echocardiography (Turner — aortic anomalies)
- Renal ultrasound (Turner — horseshoe kidney in ~10%)
Step 3 — If FSH/LH are LOW (hypogonadotropic):
- MRI brain (pituitary/hypothalamic region) — craniopharyngioma, pituitary adenoma, empty sella
- Smell test (Kallmann — formally tested with University of Pennsylvania Smell Identification Test or simple smell identification)
- GnRH stimulation test — distinguishes CDGP (LH rises to pubertal level after stimulation) from permanent hypogonadotropic hypogonadism (LH rise blunted); note this distinction is not always absolute early on
- Dynamic assessment over 6–12 months (spontaneous pubertal onset in CDGP vs no progression in Kallmann)
Step 4 — If FSH/LH are NORMAL (eugonadism):
- Pelvic ultrasound — uterus present or absent?
- MRI pelvis — MRKH (absent uterus + upper vagina), haematocolpos (imperforate hymen/vaginal atresia)
- Karyotype — AIS (46,XY with normal female external genitalia and absent uterus)
- Testosterone level (male-range in AIS)
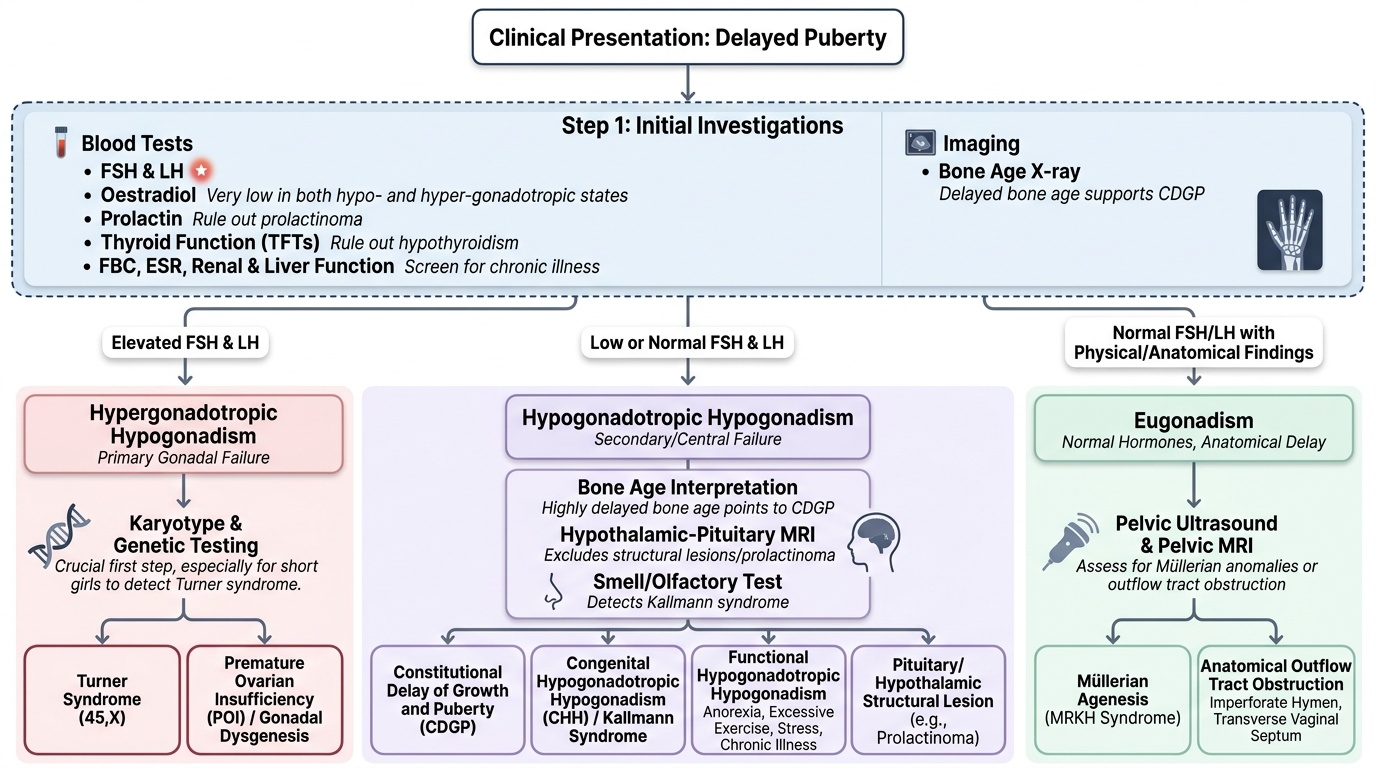
Provided image
SELF-CHECK
A 16-year-old with primary amenorrhoea has normal secondary sex characteristics (Tanner B4/PH4), normal FSH and LH, and normal oestradiol. Pelvic ultrasound shows absence of the uterus. The most likely diagnosis is:
A. Turner syndrome — karyotype will show 45,X
B. MRKH syndrome — 46,XX, absent uterus and upper vagina with normal ovaries
C. Complete androgen insensitivity syndrome — 46,XY, but needs testosterone to confirm
D. Either MRKH or complete AIS — karyotype and testosterone required to distinguish
Reveal Answer
Answer: D. Either MRKH or complete AIS — karyotype and testosterone required to distinguish
Both MRKH (46,XX) and complete AIS (46,XY) present with primary amenorrhoea, normal secondary sex characteristics, normal FSH/LH, and absent uterus. They cannot be distinguished without karyotype (MRKH = 46,XX; AIS = 46,XY) and testosterone (AIS = male-range testosterone). Turner syndrome has HIGH FSH/LH and absent secondary sex characteristics — it is excluded here.