Page 2 of 29
OG4.1 | Fetal and Placental Development — SDL Guide (Part 2)
Placental Physiology: Transfer and Endocrine Functions
The placenta performs four broad physiological roles that are operationally inseparable: gas exchange, nutrient transfer, waste elimination, and endocrine production. Understanding how each works, and at what stage of gestation each becomes critical, provides a rational framework for understanding what goes wrong in placental insufficiency.
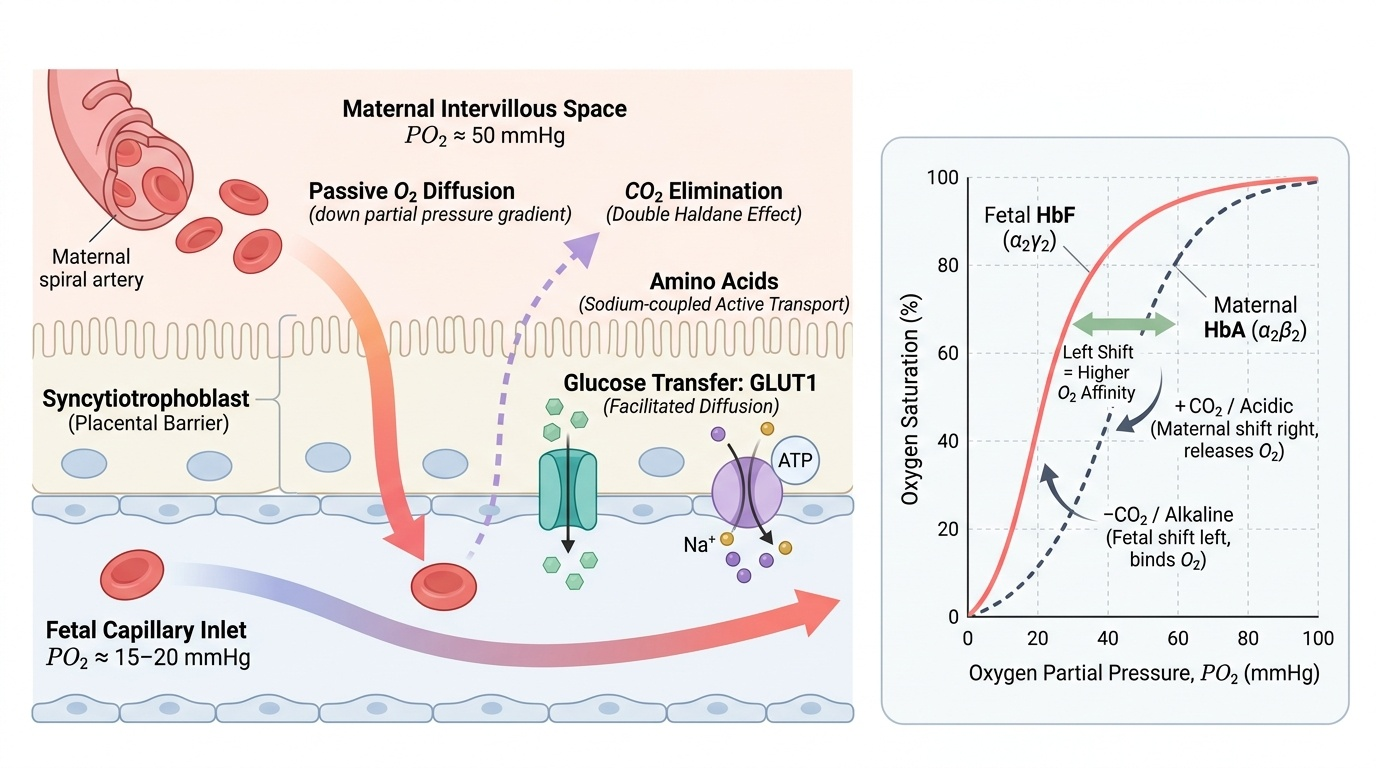
Gas exchange across the placenta is driven by partial pressure gradients and is facilitated by several mechanisms that favour fetal oxygen delivery. The partial pressure of oxygen in the intervillous space maternal blood is approximately 50 mmHg, while that in the umbilical artery (deoxygenated fetal blood arriving at the placenta) is approximately 15–20 mmHg — a gradient that drives passive diffusion of O₂ from mother to fetus. This modest gradient is made sufficient by two key physiological aids. First, fetal haemoglobin (HbF, α₂γ₂) has a higher oxygen affinity than maternal HbA (α₂β₂) because the γ-chains bind 2,3-diphosphoglycerate (2,3-DPG) poorly; the oxygen-haemoglobin dissociation curve for HbF is shifted to the LEFT, meaning HbF takes up oxygen more readily at any given PO₂. Second, the double Bohr effect amplifies this: as fetal blood releases CO₂ into the intervillous space, the maternal blood becomes more alkaline (its curve shifts right — releasing more O₂) while the fetal blood becomes more alkaline on the fetal side (its curve shifts left — picking up O₂). This elegant countercurrent exchange maximises O₂ delivery.
Carbon dioxide diffuses readily from fetal to maternal blood (CO₂ is highly diffusible), and the double Haldane effect further assists CO₂ elimination on the maternal side.
Nutrient transfer uses multiple mechanisms depending on the molecule:
- Glucose: facilitated diffusion via GLUT1 transporters on the syncytiotrophoblast; the placenta itself is a major glucose consumer, which can become clinically relevant in maternal diabetes
- Amino acids: active transport against a concentration gradient, using sodium-coupled amino acid transporters; impaired in IUGR
- Lipids: very-long-chain fatty acids (VLCFAs) and essential fatty acids transfer by diffusion; placental lipases liberate fatty acids from maternal lipoproteins
- Immunoglobulins: IgG crosses by receptor-mediated endocytosis (FcRn receptor); IgM does NOT cross; the transferred IgG provides passive neonatal immunity for the first 3–6 months
- Water: by osmosis; calcium, iron, and other minerals by active transport
Waste elimination: fetal urea, CO₂, and bilirubin cross to the maternal circulation for excretion by maternal organs.
Endocrine functions — the placenta is arguably the most prolific endocrine organ of pregnancy:
- Human chorionic gonadotrophin (hCG): a glycoprotein with α and β subunits, produced by the syncytiotrophoblast from the time of implantation. Its primary early role is to rescue the corpus luteum, maintaining progesterone production until the placenta can take over (the luteo-placental shift, at approximately 8–10 weeks). hCG levels double approximately every 48 hours in normal early pregnancy — the basis of the serial β-hCG monitoring used to confirm intrauterine location and pregnancy viability. Levels peak at 10–12 weeks of gestation, then fall to a plateau maintained throughout the second and third trimesters. hCG is the basis of all pregnancy tests.
- Human placental lactogen (hPL) — also called chorionic somatomammotropin (hCS): a polypeptide produced by the syncytiotrophoblast from approximately 6 weeks, with levels rising progressively through pregnancy. hPL acts as an insulin antagonist — it promotes maternal lipolysis (increasing free fatty acids and ketones available for fetal use) and spares glucose for the fetus. hPL stimulates mammary gland development and is the primary driver of gestational insulin resistance, the physiological basis of gestational diabetes mellitus (GDM).
- Progesterone: in the first 8–10 weeks, produced predominantly by the corpus luteum. After the luteo-placental shift, the placenta becomes the dominant source. Progesterone maintains uterine quiescence (myometrial relaxation), supports endometrial/decidual function, and suppresses T-cell-mediated immune rejection of the fetal allograft.
- Oestrogens (primarily oestriol, E₃, in pregnancy): the placenta lacks the full enzymatic complement for oestrogen synthesis from scratch; it converts DHEA-S from the fetal and maternal adrenal glands into oestrogens via placental aromatase. The measurement of unconjugated oestriol (uE₃) as a component of the quadruple screen (along with AFP, hCG, inhibin A) reflects both fetal and placental endocrine competence.
IMPORTED POINT: Comparison of placental hormones:
Provided image
IMPORTED POINT: Diagram of gas exchange mechanisms:
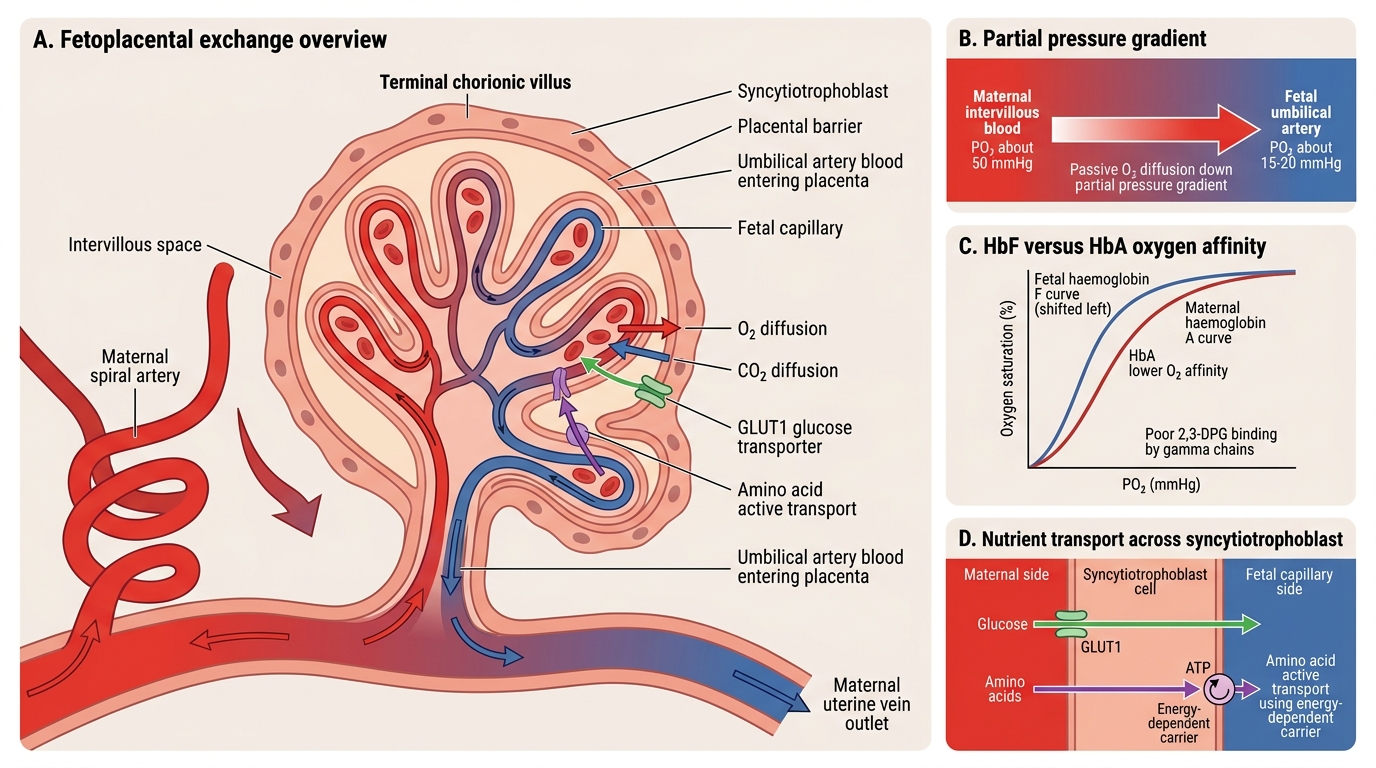
Fetoplacental Gas and Nutrient Exchange
The placental barrier — sometimes misleadingly called the haemochorial barrier — is not a true barrier in the pharmacological sense; most small, lipid-soluble, protein-unbound molecules cross freely. What determines drug transfer is molecular weight (<600 Da cross readily; >1000 Da cross poorly), lipid solubility (high = easier crossing), protein binding (only unbound drug transfers), and ionisation state (un-ionised form crosses better). This has direct implications for prescribing: for example, heparin (high MW ~15,000 Da) does NOT cross, whereas warfarin (low MW, lipid-soluble) crosses readily and causes fetal embryopathy.
SELF-CHECK
Which of the following best explains why fetal haemoglobin (HbF) is more effective than adult haemoglobin (HbA) at extracting oxygen from the placenta?
A. HbF has more haem groups per molecule, giving greater total oxygen-carrying capacity
B. HbF binds 2,3-DPG less avidly than HbA, shifting its oxygen-dissociation curve to the left and increasing O₂ affinity
C. HbF has a higher concentration in fetal blood, creating a diffusion gradient
D. HbF is more soluble, allowing it to dissolve into the intervillous space where it directly binds O₂
Reveal Answer
Answer: B. HbF binds 2,3-DPG less avidly than HbA, shifting its oxygen-dissociation curve to the left and increasing O₂ affinity
HbF (α₂γ₂) binds 2,3-DPG less avidly than HbA (α₂β₂) because the γ-subunits lack the amino acid residues that 2,3-DPG binds on β-subunits. Since 2,3-DPG reduces haemoglobin oxygen affinity (shifts curve right), its reduced binding in HbF results in a left-shifted ODC with higher oxygen affinity at any given PO₂. This allows HbF to take up oxygen from the intervillous space even at the relatively low PO₂ prevailing there. HbF has the same number of haem groups (4) as HbA — option A is wrong.
Factors Influencing Fetal Growth and Development
Fetal growth is not simply a passive process of cell multiplication. It is an active, tightly regulated biological outcome that depends on the integration of genetic information carried by the fetus, the nutritional and metabolic environment provided by the mother, the capacity of the placenta to transfer substrates and support blood flow, and a complex hormonal growth-factor axis operating in the fetus itself. Understanding each determinant individually is essential for interpreting why growth fails in a given clinical scenario — whether the problem is genetic, maternal, placental, or some combination.
1. Genetic determinants exert the strongest single influence on birth weight, accounting for approximately 40–80% of variance in population studies. This includes the fetal genome (size, metabolic rate, tissue anabolism), fetal sex (male fetuses tend to be approximately 150 g heavier than females at term), and parental stature. Genomic imprinting is particularly important in fetal growth: IGF2 (paternal imprinting — paternally expressed) drives fetal growth while H19 (maternally expressed, suppresses IGF2) restrains it. Abnormalities of imprinting cause conditions like Beckwith-Wiedemann syndrome (overgrowth) and Silver-Russell syndrome (growth restriction).
2. Maternal nutritional status is the principal modifiable determinant of fetal growth at the population level. Adequate caloric intake, protein (particularly essential amino acids), and micronutrients are all required:
- Macronutrients: severe maternal caloric restriction (as seen in famine or extreme poverty) produces symmetric IUGR. Protein restriction specifically impairs fetal organ development, with particular vulnerability of the pancreas (reduced β-cell mass, predisposing to adult Type 2 diabetes — the Barker 'thrifty phenotype' hypothesis).
- Iron: essential for haematopoiesis and myoglobin; maternal iron deficiency anaemia reduces fetal oxygen delivery and is a significant cause of IUGR in resource-limited settings.
- Folate: required for neural tube closure (critical days 22–28 post-fertilisation / weeks 3–4 of gestation); deficiency causes neural tube defects (NTDs). Folate supplementation at least one month before conception and through the first trimester reduces NTD incidence by approximately 70%.
- Iodine: required for thyroid hormone synthesis; maternal iodine deficiency causes cretinism and intellectual disability in the offspring.
- Zinc, calcium, vitamin D: contribute to skeletal mineralisation and immune development.
3. Placental factors determine the capacity for substrate delivery, independent of maternal supply:
- Surface area of the chorionic villi — placental weight correlates with villous surface area and thus exchange capacity. A small placenta (weight <10th centile for gestation) typically underlies asymmetric IUGR.
- Placental blood flow — uteroplacental blood flow at term is approximately 500–700 mL/min. Reductions impair both substrate delivery and waste removal. Conditions that reduce flow include pre-eclampsia, antiphospholipid syndrome, chronic hypertension, and severe maternal anaemia.
- Transport protein expression — GLUT1 and amino acid transporter expression on the syncytiotrophoblast is downregulated in IUGR, contributing to the growth restriction independently of blood flow.
4. Uteroplacental blood flow deserves separate emphasis. The spiral artery remodelling described earlier (invasion by cytotrophoblast cells, loss of muscular wall, conversion to wide-bore conduits) is the critical first-trimester event. In normal pregnancy, this remodelling extends into the myometrial portion of the spiral arteries by approximately 20 weeks. When remodelling is shallow — restricted to the decidual layer — the spiral arteries remain responsive to vasoconstrictive stimuli and blood flow is reduced and pulsatile. This inadequate remodelling is the primary upstream cause of pre-eclampsia and placenta-based IUGR.
5. Fetal hormones and growth factors are potent local regulators of fetal growth and organogenesis:
- IGF-1 (insulin-like growth factor-1): the primary driver of fetal growth in the second and third trimester, produced by virtually all fetal tissues. Levels correlate strongly with birth weight; IUGR fetuses have low circulating IGF-1.
- IGF-2: predominantly expressed in early gestation and in the placenta; drives placental growth and early embryonic growth. Paternally imprinted (paternal allele expressed).
- Insulin: a major fetal anabolic hormone after mid-gestation. Promotes glucose uptake, glycogen storage, protein synthesis, and adipogenesis. Fetal hyperinsulinaemia in diabetic mothers causes macrosomia (excessive growth). Pancreatic agenesis or β-cell dysfunction causes fetal growth restriction.
- Fetal thyroid hormones: essential for brain myelination, skeletal maturation, and metabolic rate. Congenital hypothyroidism (cretinism) — if untreated — causes severe intellectual disability. Maternal iodine deficiency or antithyroid antibody transfer is a correctable cause.
- Growth hormone: plays a minor role in fetal growth (the GH axis is not fully functional in utero); IGF-1 is the dominant GH-independent fetal growth signal.
6. Oxygen availability — oxygen is both a substrate and a growth signal. The fetus exists in a relatively hypoxic environment by adult standards (fetal PO₂ ~30–35 mmHg in the umbilical vein), but this is normal and HbF's high affinity ensures adequate oxygen delivery. Chronic fetal hypoxia (from high altitude, severe maternal anaemia, cardiorespiratory disease, or placental insufficiency) triggers a redistribution response — Doppler studies show increased flow to the brain (brain-sparing) and reduced flow to the gut and limbs — but sustained, prolonged hypoxia results in growth restriction and ultimately acidaemia.
7. Teratogens and timing — any agent (drug, virus, radiation, chemical) capable of disrupting normal development is a teratogen. The concept of critical periods is central: each organ system is most vulnerable during its phase of rapid differentiation.
IMPORTED POINT: Critical period diagram:
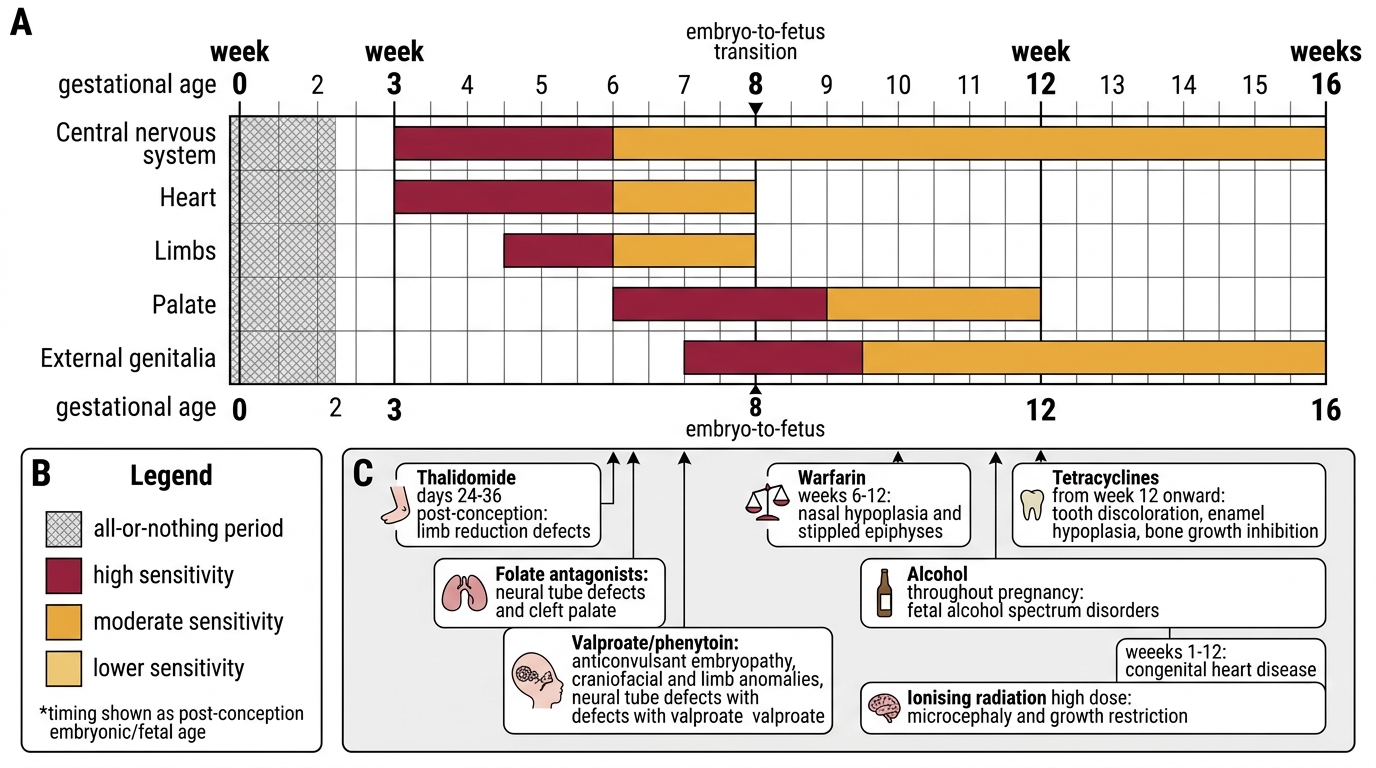
Critical Periods of Teratogen Vulnerability
IMPORTED POINT: Key teratogen examples at a glance:
- Thalidomide: weeks 24–36 post-conception → limb reduction defects (phocomelia); cautionary historical lesson
- Folate antagonists (methotrexate, trimethoprim in high dose): neural tube defects, cleft palate
- Phenytoin/valproate: fetal anticonvulsant syndrome (craniofacial, limb anomalies, NTDs for valproate)
- Warfarin: embryopathy (nasal hypoplasia, stippled epiphyses) at 6–12 weeks; fetal haemorrhage throughout
- Tetracyclines: weeks 12 onward → dental discolouration and enamel hypoplasia, bone growth inhibition
- Rubella virus: weeks 1–12 (first trimester) → classic triad of congenital rubella — cataracts, sensorineural deafness, congenital heart disease (PDA, pulmonary stenosis)
- Alcohol: throughout pregnancy — no safe dose identified; fetal alcohol spectrum disorders (facial dysmorphia, growth restriction, intellectual disability)
- Ionising radiation: >50 mGy in embryonic period → microcephaly, growth restriction; diagnostic doses are far below this threshold
8. Maternal disease can impair fetal growth through multiple mechanisms:
- Hypertension/pre-eclampsia → reduced uteroplacental blood flow
- Diabetes mellitus → IGF-1 and insulin stimulation → macrosomia (type 1/GDM, poorly controlled) OR asymmetric IUGR if associated with hypertension/microangiopathy
- Chronic kidney disease → combined effect of hypertension, anaemia, and protein loss
- Antiphospholipid syndrome → placental thrombosis → IUGR and recurrent miscarriage
- Severe systemic lupus → anti-Ro/anti-La antibodies → fetal heart block; placental disease → IUGR
IMPORTED POINT: Organising mental model of fetal growth determinants:
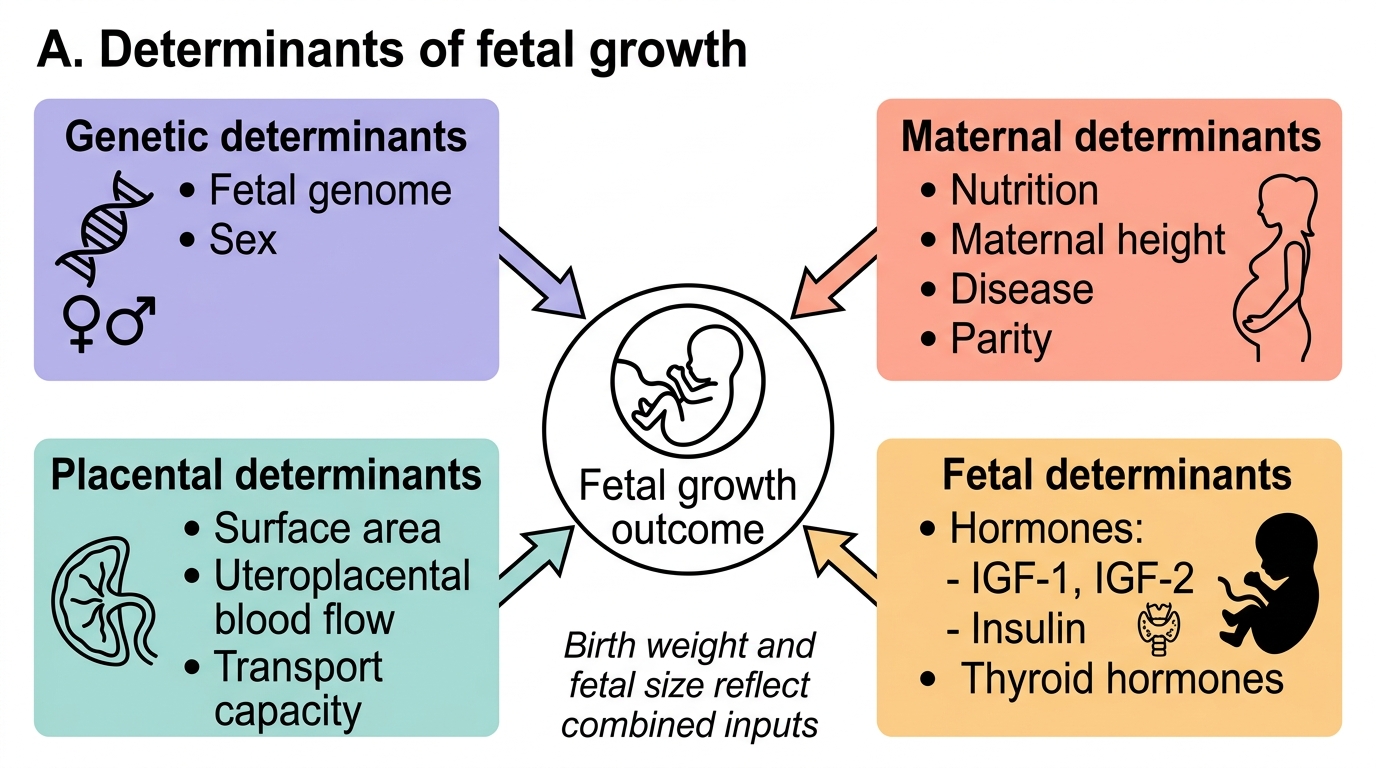
Determinants of Fetal Growth
CLINICAL PEARL
The double Bohr effect at the placenta is clinically underestimated. When students learn that fetal PO₂ is only ~30 mmHg, they often wonder how the fetus manages to extract adequate oxygen. The answer lies in three simultaneous mechanisms: (1) HbF's left-shifted oxygen dissociation curve (due to poor 2,3-DPG binding), (2) the double Bohr effect — fetal CO₂ release into the intervillous space makes maternal HbA unload more O₂ (Bohr shift right) while the fetal HbF simultaneously shifts left, enhancing uptake, and (3) the Haldane effect — CO₂ is carried more efficiently as carbamino-haemoglobin by deoxygenated blood, assisting CO₂ removal from the fetus. These three mechanisms work in concert, and it is their combination — not just HbF alone — that makes fetal gas exchange viable despite the apparently unfavourable partial pressures.
Clinical Significance: From Basic Science to Bedside
The clinical applications of fetal and placental developmental biology span the entire antenatal period and extend into neonatal care. This section traces the direct lines from the basic science to clinical practice — illustrating why the knowledge from the previous sections is not decorative but operationally essential.
1. Fetal dating and growth surveillance rest entirely on knowledge of normal developmental milestones. Gestational age assignment at the 11–13 week scan uses the crown-rump length (CRL) because this measurement has the smallest biological variability at this gestation and is the most accurate dating method available. Beyond 14 weeks, the biparietal diameter (BPD), head circumference (HC), abdominal circumference (AC), and femur length (FL) — all derived from knowledge of normal fetal growth — are used on serial scans to assess growth and detect the small fetus. A fetus with an AC falling below the 10th centile on a customised growth chart triggers investigation for fetal growth restriction (FGR).
2. Intrauterine growth restriction (IUGR / FGR) is the clinical syndrome in which the fetus fails to achieve its growth potential. Early-onset IUGR (before 32 weeks) is most commonly due to severe placental insufficiency with impaired spiral artery remodelling — the pathological correlate of what we described in the growth determinants section. The Doppler finding of absent or reversed end-diastolic flow (AREDF) in the umbilical artery signals extreme placental resistance and impending fetal compromise. Late-onset FGR (after 32 weeks) is more heterogeneous, including constitutional small-for-gestational-age fetuses and mildly under-perfused placentas. Management requires serial Doppler surveillance, steroids for lung maturation if delivery before 34 weeks is anticipated, and timely delivery — decisions that are rational only when the clinician understands the underlying placental pathophysiology.
3. Placental pathology — placenta praevia, in which the placenta is implanted over or near the internal cervical os, arises from abnormal implantation site selection in the blastocyst stage, possibly influenced by endometrial scarring or uterine anomaly. Understanding the normal anatomy of implantation and decidualisation helps explain why prior caesarean scars predispose to placenta praevia and placenta accreta spectrum (PAS) — in which the decidua basalis is deficient, allowing abnormal trophoblast invasion into the myometrium (accreta), perimetrium (increta), or adjacent organs (percreta). The pathophysiology of PAS is a direct extension of the normal trophoblast invasion mechanisms described earlier.
4. Drug transfer across the placenta has direct therapeutic implications. Drugs with low molecular weight, high lipid solubility, and low plasma protein binding cross freely. Heparin (MW ~15,000 Da) does not cross, making it the anticoagulant of choice in pregnancy. Low molecular weight heparins (LMWH) similarly do not cross at therapeutic doses. Warfarin (MW 308 Da, lipid-soluble) crosses and causes warfarin embryopathy (6–12 weeks) and fetal haemorrhage at any gestation. Magnesium sulphate crosses the placental barrier; neonatal hypermagnesaemia with respiratory depression can occur when the maternal MgSO₄ dose is not stopped before delivery. Corticosteroids (betamethasone/dexamethasone) given to the mother for fetal lung maturation must cross the placenta — and they do, because they are lipid-soluble fluorinated steroids; prednisolone and methylprednisolone are largely metabolised by the placenta and do NOT reach the fetus in significant concentrations.
5. Rh sensitisation occurs when Rh-positive fetal red blood cells (RBCs) enter the circulation of an Rh-negative mother, triggering the formation of anti-D IgG antibodies. This can occur at delivery, termination, amniocentesis, external cephalic version, antepartum haemorrhage, or spontaneously. In a subsequent Rh-positive pregnancy, maternal anti-D IgG crosses the placenta (via FcRn-mediated endocytosis, as described in the nutrient transfer section) and destroys fetal RBCs, causing haemolytic disease of the newborn (HDN). Prophylactic anti-D immunoglobulin works by clearing fetal RBCs from the maternal circulation before B-cell memory is established. The entire mechanism of sensitisation and prophylaxis is grounded in the anatomy of the fetoplacental barrier and the IgG transfer mechanism.
6. Anomaly screening at 11–13 weeks (nuchal translucency + maternal serum markers) and 18–20 weeks (morphology scan) is underpinned by knowledge of when each fetal structure forms and when it is visible. For example, the four-chamber cardiac view is assessable from 18 weeks because cardiac septation is complete by 8 weeks — any defect present at 18 weeks is a structural remnant of a failed embryonic process. Similarly, the nuchal fold thickens in Down syndrome because of lymphatic drainage failure — a consequence of a chromosomal abnormality affecting lymphatic development during the embryonic period.
7. Fetal lung maturity is assessed clinically at borderline gestations (between 34 and 37 weeks). Antenatal corticosteroids accelerate surfactant production in the type II pneumocytes of the fetal lung — the same cells that begin surfactant synthesis from approximately 24 weeks. The timing of steroid administration is calibrated to the maturation timeline of the fetal lung.
SELF-CHECK
A 30-year-old Rh-negative woman with a first pregnancy delivers an Rh-positive baby. She was not given prophylactic anti-D immunoglobulin postpartum. In her second Rh-positive pregnancy, the baby develops severe haemolytic disease of the newborn (HDN). Which step in the pathophysiology of HDN directly depends on a specific placental transport mechanism discussed in this module?
A. Formation of anti-D IgM antibodies in the mother during the first pregnancy
B. Passive transfer of maternal anti-D IgG across the placenta to the fetus via FcRn receptor-mediated endocytosis
C. Release of fetal red blood cells into the maternal circulation during labour
D. Destruction of fetal RBCs in the fetal spleen after antibody binding
Reveal Answer
Answer: B. Passive transfer of maternal anti-D IgG across the placenta to the fetus via FcRn receptor-mediated endocytosis
The step that depends on a placental mechanism is the transfer of maternal anti-D IgG across the placental barrier. As described in the nutrient transfer section, IgG — unlike IgM — crosses the placenta via receptor-mediated endocytosis using the neonatal Fc receptor (FcRn) on the syncytiotrophoblast. IgM cannot cross because of its large pentameric structure (MW ~900,000 Da) and lack of FcRn affinity. In the first pregnancy, IgM is produced (does not reach the fetus significantly) and then memory B cells form IgG. In the second pregnancy, the IgG readily crosses via FcRn and destroys fetal RBCs. Option A (IgM formation) and option C (fetoplacental haemorrhage) are part of the sequence but not specifically placental transport mechanisms.
Key Concepts for Self-Assessment
This section summarises the core self-test points from the module and is designed for active self-directed review before you proceed to assessments or clinical sessions. Competency OG4.1 sets a Knowledge-Discuss (KH) expectation — you are expected not merely to recall individual facts but to reason with them, linking embryological events to clinical consequences, explaining physiological mechanisms rather than listing them, and integrating the determinants of fetal growth into a coherent framework. The questions below are organised into four thematic domains that mirror the arc of the module: embryology and fetal milestones; placental anatomy; placental physiology; and factors influencing fetal growth. As you work through each question, challenge yourself to explain the underlying mechanism, not simply state the fact. If a question reveals a gap — a milestone date you are uncertain of, a hormone peak you cannot recall, a Doppler finding whose physiology is blurred — return to the relevant section and read the lead prose again before re-testing. This iterative approach is the essence of self-directed learning and is what this module is designed to support.
Embryology and fetal milestones:
- What is the boundary between the embryonic and fetal periods, and why does it matter clinically?
- At what gestational week does implantation begin and end? What layers does the trophoblast differentiate into at implantation?
- Which weeks of gestation constitute the highest risk period for structural teratogens, and which organ systems are most vulnerable in which weeks?
- What is the 'all-or-nothing' period and what is its biological explanation?
- State the approximate CRL and weight at 12, 20, 28, and 40 weeks.
Placental anatomy:
- Distinguish the syncytiotrophoblast from the cytotrophoblast: which layer is which, what are the functional roles, and how do they change across gestation?
- Trace the path of fetal blood through the placenta, naming vessels and structures from the umbilical artery to the umbilical vein.
- What is the intervillous space, and what does it contain?
- How many vessels are in a normal umbilical cord, and what does a single umbilical artery (SUA) signify?
Placental physiology:
- Why does HbF load oxygen more readily than HbA at the PO₂ prevailing in the placenta?
- Name the four main placental hormones, state when each peaks in gestation, and give one clinical application of each.
- When does the luteo-placental shift in progesterone production occur, and what are the consequences if it fails?
- Name two drug classes that DO cross the placenta and two that do NOT, explaining the molecular basis in each case.
Factors influencing fetal growth:
- List the four broad categories of fetal growth determinants with two examples each.
- Which growth factor is most strongly correlated with third-trimester fetal weight? Which paternal-imprinting locus promotes fetal growth?
- What is the physiological basis by which insulin drives macrosomia in poorly controlled gestational diabetes?
- At what gestational week do spiral artery remodelling events occur, and what clinical complication is associated with shallow remodelling?