Page 1 of 13
OP5.1 | Episcleritis — SDL Guide
Learning Objectives
- Describe the etiopathogenesis of episcleritis and scleritis, including the role of immune-mediated inflammation
- Classify episcleritis (simple, nodular) and scleritis (anterior: diffuse, nodular, necrotising; posterior) using the Watson & Hayreh scheme
- Identify the key clinical features that distinguish episcleritis from scleritis, including the phenylephrine 2.5% blanching test
- Outline the complications of untreated or severe scleritis
- Describe the management of episcleritis and the principles of management of scleritis
INSTRUCTIONS
Episcleritis and scleritis both present as red eye but differ fundamentally in depth, severity, and consequence. Episcleritis is one of the commonest causes of a red eye in young adults and is almost always benign and self-limiting. Scleritis, by contrast, is a vision-threatening ocular emergency with strong associations with systemic vasculitic and connective tissue diseases. Mastering the clinical distinction — especially the phenylephrine blanching test — allows you to correctly stratify patients, avoid over-investigating a benign condition, and never miss a sight-threatening one.
References
- Khurana AK. Comprehensive Ophthalmology. 7th ed. New Delhi: New Age International, 2023. Ch 6 (Diseases of the Sclera) (textbook)
- Parsons' Diseases of the Eye. 22nd ed. Philadelphia: Elsevier, 2011. Chapter on Diseases of the Sclera (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 28-year-old medical student walks into the eye OPD with a three-day history of redness in the left eye. She reports mild aching discomfort but no severe pain, no discharge, and her vision feels perfectly normal. On inspection the redness is sectoral — confined to the temporal quadrant — and the vessels look bright red and superficial. She is anxious: her roommate was recently diagnosed with rheumatoid arthritis after presenting with a similar-looking red eye. You look at the eye under the slit-lamp and wonder: is this a benign self-limiting condition that needs only reassurance, or is this the beginning of something that could destroy her vision?
You instil a drop of phenylephrine 2.5% and wait ninety seconds. The sectoral redness blanches completely. You breathe more easily — this is episcleritis, not scleritis. But you know that distinguishing the two requires more than just one drop of eye medication.
WHY THIS MATTERS
Episcleritis is the single commonest cause of acute unilateral red eye in outpatient ophthalmology practice. It affects primarily young adults and women, has no clear trigger in most cases, and almost always resolves spontaneously within two to three weeks. Yet the same anatomical region — the episcleral tissue covering the sclera — can also harbour scleritis, a condition that is sight-threatening, intensely painful, and frequently the presenting sign of a serious systemic vasculitis such as granulomatosis with polyangiitis (formerly Wegener's granulomatosis) or rheumatoid arthritis. Missing scleritis and treating it as simple episcleritis can lead to progressive scleral necrosis, corneal melt, and even globe perforation. Conversely, labelling every case of episcleritis as scleritis leads to unnecessary systemic work-up, harmful steroid courses, and patient anxiety. The phenylephrine blanching test and careful slit-lamp examination are your primary tools to make this critical distinction at the bedside — a skill that every MBBS graduate must master.
RECALL
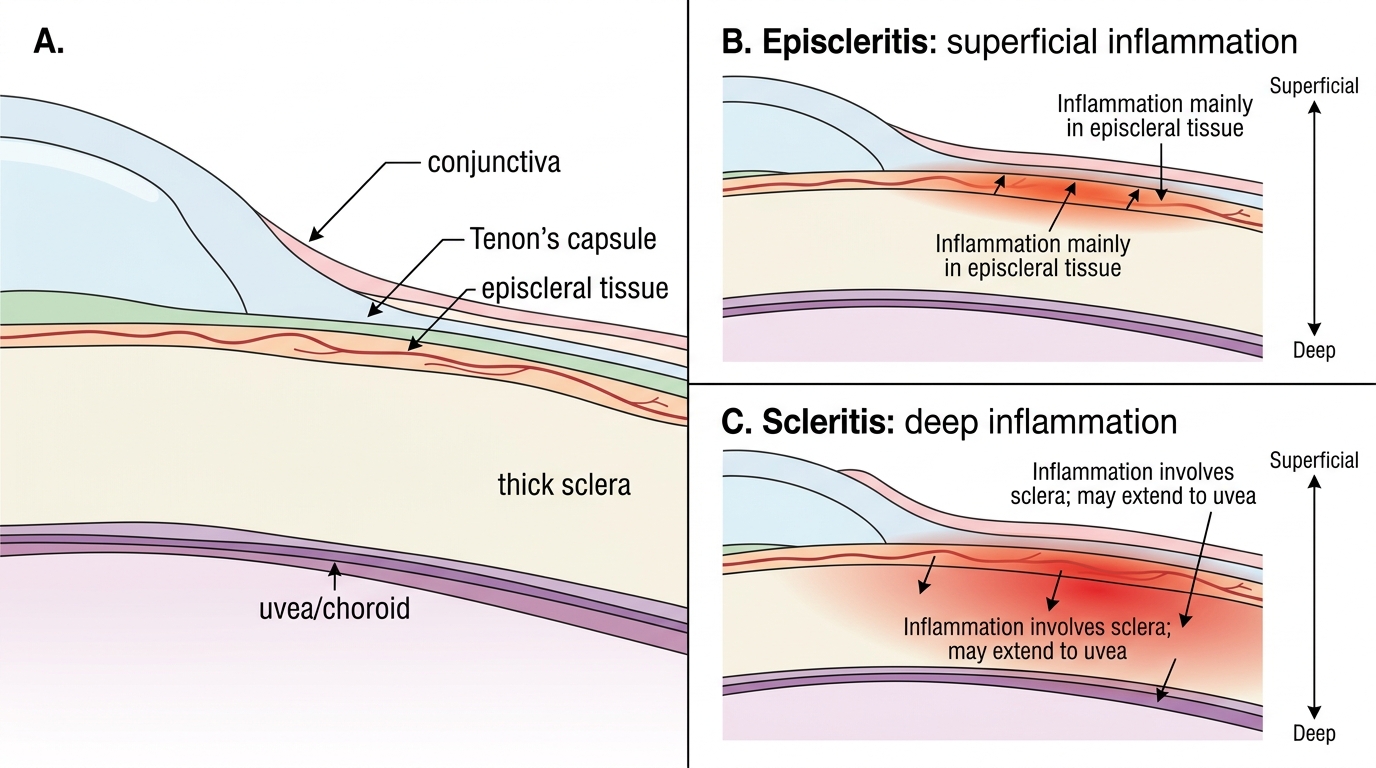
Before exploring the pathology, recall the ocular surface anatomy. The outermost visible layer is the conjunctiva — a thin transparent mucous membrane that lines the inner eyelids (palpebral) and covers the anterior sclera (bulbar conjunctiva). Deep to the conjunctiva lies Tenon's capsule (the fascia bulbi), and between Tenon's capsule and the scleral surface is a thin layer of loose connective tissue called the episclera. The sclera itself is a dense, relatively avascular, collagen-rich white coat that gives the eye its structural rigidity. The episcleral tissue contains a rich plexus of fine blood vessels; the sclera proper has very sparse vascularity in its outer layers and is essentially avascular centrally. This anatomical layering is the key to understanding why episcleral inflammation (episcleritis) produces a superficial, bright-red appearance that blanches, whereas scleral inflammation (scleritis) produces a deep, violaceous (bluish-red) hue that does not blanch. You also recall from pathophysiology that immune-mediated inflammation driven by complement activation, immune-complex deposition, and T-lymphocyte infiltration is the basis for both conditions — explaining their strong associations with autoimmune disease.
Clinical Presentation of Episcleritis
Episcleritis is the most common cause of acute red eye in young to middle-aged adults, with a peak incidence between 20 and 50 years and a slight female preponderance. The presentation is characteristically mild and unilateral in onset, though bilateral involvement occurs in up to a third of cases. The patient reports a sudden or subacute onset of sectoral redness — a wedge-shaped area of bright-red injection typically in the interpalpebral zone (the temporal or nasal quadrant), though it can be more diffuse. The discomfort is described as mild-to-moderate aching or a sense of ocular irritation; it is distinctly NOT the severe, boring, nocturnal pain that characterises scleritis. There is usually no significant photophobia, no discharge, and visual acuity is fully preserved. Tearing may be present.
Episcleritis occurs in two clinical subtypes. Simple episcleritis is the more common form (approximately 70% of cases), presenting as a diffuse sector of episcleral injection that is bright red. It tends to be recurrent, with episodes lasting two to three weeks each. Nodular episcleritis accounts for the remainder and presents with a discrete, slightly raised, tender, mobile nodule of inflamed episcleral tissue, again typically in the interpalpebral zone. The nodule is localised and can be moved slightly with a cotton-tipped swab (unlike scleral nodules which are fixed). Recurrences in nodular episcleritis are more common and episodes tend to be more prolonged.
In the vast majority of cases — roughly 70% — episcleritis is idiopathic, with no identifiable systemic cause. When a systemic association is found it is most commonly with inflammatory bowel disease, rosacea, gout, atopic conditions, and occasionally the same conditions that cause scleritis (rheumatoid arthritis, vasculitis). This is why a careful history including any joint symptoms, skin rashes, bowel symptoms, and prior episodes is always warranted on the first presentation.
Clinical Presentation of Scleritis
Scleritis is a far less common but far more serious condition than episcleritis. The cardinal symptom that distinguishes scleritis from every other cause of red eye is the character and severity of pain: patients describe a severe boring or aching pain that is deep, persistent, and characteristically worse at night — often waking the patient from sleep. The pain is typically periocular, may radiate to the forehead, temple, or jaw (due to involvement of the ophthalmic branch of the trigeminal nerve), and is aggravated by eye movement. Tenderness on gentle palpation of the globe through the closed eyelid is a consistent and important sign — pressure over the inflamed scleral area reproduces the pain. Visual acuity may be reduced in more severe forms due to involvement of the cornea, uvea, or posterior segment.
The eye appears diffusely or sectorally red with a characteristic violaceous (bluish-red) hue, reflecting the deep congestion of scleral vessels. Unlike the bright-red superficial injection of episcleritis, the deep redness of scleritis does not shift on applying gentle pressure or blanch with phenylephrine. Slit-lamp examination reveals the injection to arise from the deep episcleral plexus and scleral vasculature.
Scleritis is strongly associated with systemic autoimmune disease in approximately 50% of patients. Rheumatoid arthritis is the commonest association, present in 20-30% of scleritis patients, particularly in the necrotising form. Other important systemic associations include granulomatosis with polyangiitis (GPA/Wegener's granulomatosis), relapsing polychondritis, systemic lupus erythematosus (SLE), Crohn's disease, and ankylosing spondylitis. Because scleritis may be the presenting manifestation of an occult systemic vasculitis, a thorough systemic review and appropriate investigations are mandatory once the diagnosis of scleritis is established.
Anatomy and Pathophysiology of Episcleritis and Scleritis
To understand why episcleritis and scleritis behave so differently clinically, one must appreciate the anatomical layering of the ocular wall and the nature of the inflammatory process in each tissue compartment. The episclera is a thin layer of loose, vascular connective tissue that lies between Tenon's capsule (the fibrous sheath of the eyeball) and the outer surface of the sclera. It contains a rich superficial vascular plexus. The sclera itself is composed of dense, randomly interwoven collagen fibres with relatively sparse cellularity and minimal intrinsic vasculature — its nutrition comes mainly from the superficial episcleral vessels and the deeper uveal circulation. This fundamental difference in vascular density and tissue laxity, combined with the differing immunological microenvironments of the two tissue planes, is why:
- Episcleral inflammation is vascular, exudative, and usually self-limiting (rich vessels → brisk response but also brisk resolution; lax tissue → exudate drains easily).
- Scleral inflammation is deep, poorly-vascularised, and destructive — the avascular collagen matrix cannot mount an efficient resolution response, so immune-complex deposition and cytokine-mediated collagen degradation can progress unimpeded.
The pathophysiology of both conditions is fundamentally immune-mediated. In episcleritis, the inflammatory infiltrate is predominantly neutrophilic in the acute phase, with episcleral oedema and vascular engorgement; T-lymphocyte infiltration is modest and complement activation is limited. The absence of immune-complex deposition in the scleral layers explains the benign course. In scleritis, particularly the diffuse and necrotising forms, the pathology shows granulomatous infiltration with epithelioid macrophages and giant cells, immune-complex deposition in vessel walls, complement activation, and vasculitis — a process that closely mirrors the systemic vasculitis seen in RA and GPA. The resulting collagen destruction can thin the sclera to a translucent bluish staphyloma (uveal tissue visible through the thinned sclera) and ultimately to perforation.
Etiopathogenesis of idiopathic episcleritis remains incompletely understood; a localised Type III hypersensitivity (immune-complex) reaction in the episcleral vessels has been proposed, though many cases may represent a forme fruste of a broader immune activation triggered by remote infection, stress, or subclinical systemic disease. For scleritis, immune-complex vasculitis and cell-mediated (Type IV) hypersensitivity are both operative, explaining why systemic immunosuppression is necessary for severe cases.
Inflammation Depth in Episcleritis and Scleritis