Page 5 of 13
OP5.2 | Systemic Associations in Episcleritis/Scleritis Referral — SDL Guide
Learning Objectives
- Enumerate the systemic conditions associated with episcleritis and scleritis
- Explain why scleritis — more than episcleritis — is associated with severe systemic vasculitides
- Identify clinical clues in the history and examination that suggest an underlying systemic disease
- Outline the investigative panel required for a new patient with confirmed scleritis
- State the indications for referral in episcleritis and scleritis, specifying the appropriate specialist
INSTRUCTIONS
The ophthalmologist who encounters a patient with scleritis occupies a pivotal diagnostic position: the eye may be the first organ to declare a life-threatening systemic vasculitis. Up to half of all patients with scleritis have an underlying connective tissue disease or vasculitis — and in many, the ocular presentation precedes the systemic diagnosis by months or years. Understanding which systemic conditions drive ocular inflammation, how to look for them in the history and examination, and when and to whom to refer is a core competency for every MBBS graduate entering clinical practice.
References
- Khurana AK. Comprehensive Ophthalmology. 7th ed. New Delhi: New Age International, 2023. Ch 6 (Diseases of the Sclera) (textbook)
- Parsons' Diseases of the Eye. 22nd ed. Philadelphia: Elsevier, 2011. Chapter on Diseases of the Sclera (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 52-year-old man presents to the eye emergency department with a 10-day history of severe, boring right eye pain that wakes him from sleep at night. His wife insists he has 'just a red eye' and has been applying over-the-counter antibiotic drops with no benefit. On examination the eye is deeply injected with a violaceous hue, exquisitely tender on palpation, and phenylephrine 2.5% fails to blanch the injection. You diagnose anterior scleritis. You then look more carefully at the patient: he has a subtle saddle-nose deformity, a history of two episodes of epistaxis in the past year, and recently completed a course of antibiotics for a chest infection that 'wouldn't go away.' Your ANCA panel comes back: c-ANCA strongly positive. This patient does not have a red eye. He has granulomatosis with polyangiitis (GPA), and without prompt systemic treatment — cyclophosphamide and high-dose corticosteroids — his kidneys are at risk of irreversible damage. The referral you make from the eye clinic to nephrology and rheumatology that afternoon may save his life.
WHY THIS MATTERS
Episcleritis and scleritis are not merely ocular conditions — they are windows into the immune system's broader state. For the clinician, recognising the systemic associations is consequential in two directions. In scleritis, approximately 50% of patients harbour an underlying systemic connective tissue disease or vasculitis, and in a significant proportion of these, the ocular inflammation is the first overt manifestation of a condition that — if undetected — will progress to renal failure, pulmonary haemorrhage, or systemic vasculitic crisis. The ophthalmologist's diagnosis and timely referral can make the difference between organ-sparing treatment and irreversible systemic damage. Conversely, in episcleritis, systemic associations are present in roughly 30% of cases but are generally less severe — and overzealous investigation of every episode of simple episcleritis wastes resources and increases patient anxiety. Knowing when to investigate and when to reassure is as important as knowing what to investigate. Both skills — enumeration of systemic associations and clinical judgement about referral thresholds — are tested in every ophthalmology clinical examination at the MBBS and postgraduate levels.
RECALL
From the previous module (OP5.1), recall the fundamental distinction between episcleritis (superficial episcleral inflammation, self-limiting, benches with phenylephrine 2.5%, mild symptoms) and scleritis (deep scleral inflammation, severe boring pain, non-blanching, vision-threatening). The pathophysiology of scleritis established that the process is a scleral vasculitis — immune complex deposition in the scleral and episcleral blood vessel walls, with complement activation, T-lymphocyte-mediated granulomatous infiltration, and cytokine-driven collagen destruction. This is the same class of immune-complex vasculitis that drives the synovitis of rheumatoid arthritis, the granulomatous inflammation of GPA, and the vascular damage of SLE. The mechanistic link between the scleral vasculitis and the systemic vasculitides is not incidental — the sclera is merely the site where the same immune attack happens to be visible to the clinician.
Systemic Associations of Episcleritis
Episcleritis is idiopathic in approximately 70% of cases, and when a systemic association is identified, it is generally with a milder spectrum of inflammatory or immune conditions compared to scleritis. The systemic work-up for a first episode of simple episcleritis is not routinely warranted unless symptoms are atypical, the course is particularly severe or recurrent, or there are associated systemic symptoms.
It is worth emphasising to patients that an episcleritis diagnosis does not, by itself, mandate a battery of systemic investigations — the diagnostic yield is low in uncomplicated simple episcleritis, and unnecessary tests cause anxiety without clinical benefit. However, when clinical triggers are present (recurrence, bilaterality, nodular morphology, or associated systemic symptoms), a targeted screen is appropriate and may reveal a treatable underlying condition.
The systemic conditions associated with episcleritis include:
- Inflammatory bowel disease (IBD) — both Crohn's disease and ulcerative colitis are associated with episcleritis. Ocular manifestations (including episcleritis and anterior uveitis) occur in up to 10% of IBD patients and may flare in parallel with intestinal disease activity.
- Rosacea — a common dermatological condition associated with episcleral inflammation and, when involving the eye (ocular rosacea), can also cause meibomian gland dysfunction and marginal keratitis. Telangiectasias and flushing of the malar and nasal skin may be visible on examination.
- Gout — urate crystal deposition in the episcleral tissue is the proposed mechanism. Episcleritis may accompany or precede a gouty attack. Serum uric acid is a useful test in patients with recurrent episodes.
- Atopic disease — atopic dermatitis and allergic conditions have been associated with episcleral inflammation, possibly through shared mast-cell and IgE-mediated pathways.
- Rheumatoid arthritis — RA is primarily associated with scleritis (particularly necrotising forms), but mild episcleral involvement can also occur in RA patients, especially during systemic flares.
- Systemic lupus erythematosus (SLE) — episcleral involvement is less common than scleritis in SLE but has been reported.
- Herpes zoster ophthalmicus — viral reactivation can trigger segmental episcleral inflammation as part of the ocular involvement.
In clinical practice, the yield of systemic investigation in a first episode of uncomplicated episcleritis is low. Targeted investigation is justified when: the episode is particularly severe or atypical; recurrences are frequent; there are systemic symptoms suggesting IBD (diarrhoea, rectal bleeding), gout (joint pain, hyperuricaemia), or a connective tissue disease; or the phenylephrine test is equivocal and scleritis cannot be excluded.
Systemic Associations of Scleritis
Scleritis has far more powerful and clinically significant systemic associations than episcleritis. In landmark case series (notably Sainz de la Maza et al. 1994), approximately 50% of patients with scleritis have an identifiable underlying systemic disease. The conditions are dominated by connective tissue diseases and systemic vasculitides — conditions where immune-complex deposition in vessel walls and granulomatous inflammation are central mechanisms, mirroring the scleral pathology.
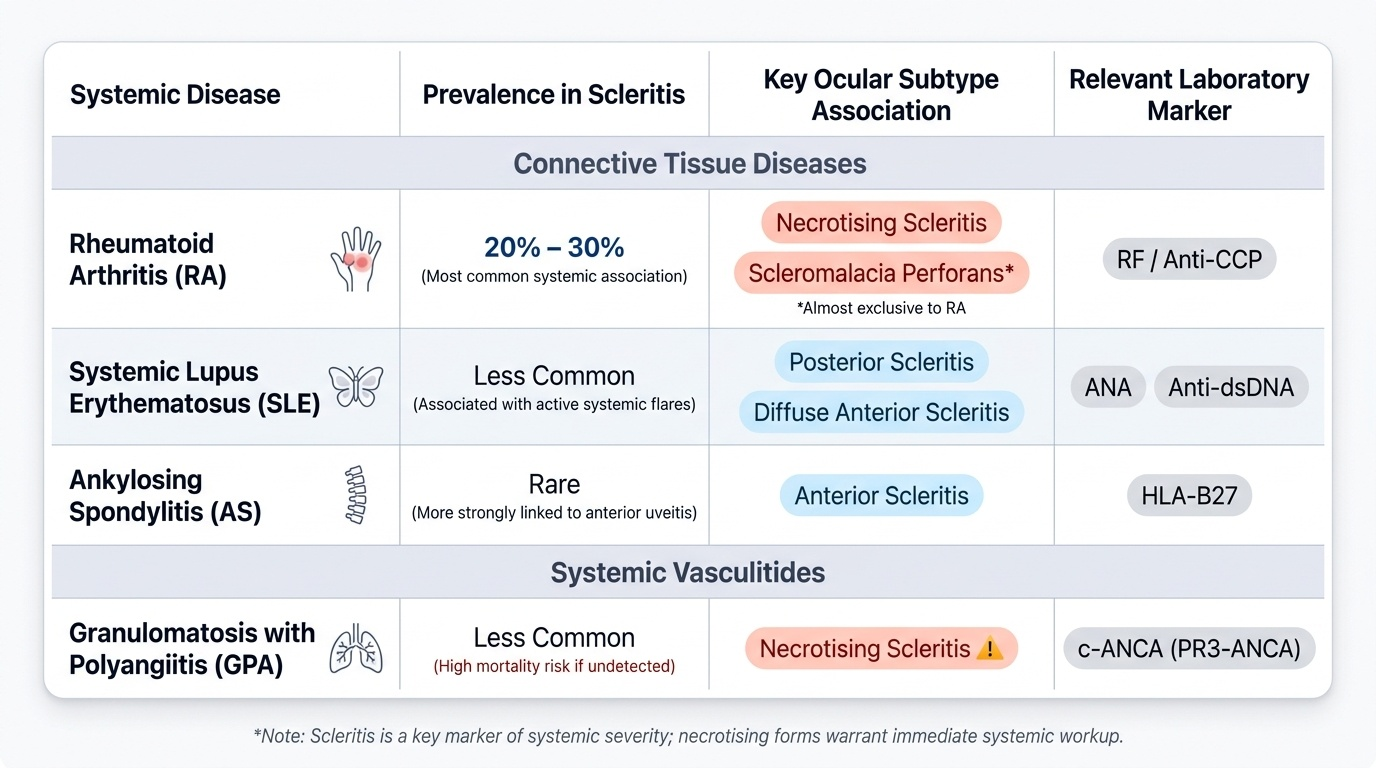
Provided image
The pattern of systemic disease in scleritis reflects a fundamental truth: scleritis is not merely an ocular condition — it is, in a substantial proportion of cases, an ophthalmological manifestation of systemic vasculitis or autoimmune connective tissue disease. This has a direct clinical implication: every new diagnosis of scleritis must trigger a systemic disease screen, because the underlying condition may carry greater mortality risk than the scleritis itself — granulomatosis with polyangiitis can cause fatal renal or respiratory failure if unrecognised. The major systemic associations, grouped by disease category, are listed below.
Connective tissue diseases:
1. Rheumatoid arthritis (RA) — the single most common systemic association, present in 20-30% of patients with scleritis. RA-associated scleritis tends to affect older women with long-standing, erosive, seropositive disease. All subtypes of scleritis are seen, but necrotising scleritis and scleromalacia perforans have the strongest RA association. Scleromalacia perforans (painless necrotising scleritis) occurs almost exclusively in RA.
2. Systemic lupus erythematosus (SLE) — ANA and anti-dsDNA antibodies; scleritis (particularly posterior and diffuse anterior) occurs in SLE, often in the context of active systemic flares with nephritis or serositis.
3. Ankylosing spondylitis (AS) — HLA-B27 associated; primarily associated with anterior uveitis, but scleritis is reported.
Systemic vasculitides:
4. Granulomatosis with polyangiitis (GPA, formerly Wegener's granulomatosis) — a necrotising granulomatous vasculitis affecting the upper respiratory tract (nasal/sinus destruction, saddle-nose deformity), lungs (cavitating nodules, haemorrhage), and kidneys (crescentic glomerulonephritis). c-ANCA (PR3-ANCA) is the serological marker. Scleritis — particularly necrotising scleritis — may be the first manifestation of GPA. Missing GPA means missing treatable renal failure.
5. Relapsing polychondritis — an autoimmune condition causing episodic inflammation of cartilaginous structures (ear pinna, nasal cartilage, tracheal rings, intervertebral discs). The ear becomes red, swollen, and painful; the nasal bridge collapses. Scleritis is one of the commonest ocular manifestations and may be the presenting feature.
6. Polyarteritis nodosa (PAN) — a systemic necrotising vasculitis affecting medium-sized vessels; scleritis is an uncommon but recognised manifestation.
7. Microscopic polyangiitis — p-ANCA (MPO-ANCA) associated; predominantly a renal and pulmonary vasculitis; scleritis reported.
Other associations:
8. Inflammatory bowel disease — IBD is associated with both episcleritis and scleritis; active IBD activity may drive ocular flares.
9. Infective causes — less commonly, scleritis can be caused by direct infection (bacterial — particularly Pseudomonas and Staphylococcus after trauma or surgery; viral — herpes zoster, syphilis). Infectious scleritis is not immune-mediated and requires antimicrobial rather than immunosuppressive therapy. A history of recent surgery, trauma, or contact lens use should always be sought.
Why Scleritis Has Such Strong Systemic Links: Pathogenic Mechanisms
The mechanistic connection between scleritis and systemic vasculitis is not coincidental — it is the same disease process operating in different anatomical sites. Understanding this link helps the clinician appreciate why investigating and treating the systemic disease is as important as managing the ocular inflammation.
The sclera, as noted in OP5.1, is a poorly-vascularised dense collagen matrix. However, its outer episcleral and anterior ciliary vessels are part of the systemic circulation and are exposed to the same circulating immune complexes, autoantibodies, and inflammatory cytokines that drive systemic vasculitis. In rheumatoid arthritis, immune complexes (IgM anti-IgG rheumatoid factor + complement) deposit in vessel walls and trigger a necrotising vasculitis in the synovium — and in the scleral vessels. The same TNF-alpha, IL-1, and IL-6 cytokines that erode cartilage in the joint also drive scleral collagen degradation. In GPA, the granulomatous T-cell infiltration that destroys the nasal septum and renal glomeruli also infiltrates the scleral stroma. This is why systemic immunosuppression (not just local eye drops) is the definitive treatment for scleritis associated with vasculitis — and why treating the eye alone without treating the underlying disease is fundamentally inadequate.
Episcleritis, by contrast, involves the loose vascular episcleral connective tissue rather than the dense scleral collagen matrix. Its pathology is predominantly a localised vascular response (likely Type III hypersensitivity in the episcleral vessels) rather than a granulomatous vasculitis — explaining why it responds to conservative treatment and has less severe systemic associations.