Page 1 of 20
OP7.1 | Congenital Glaucoma — SDL Guide
Learning Objectives
- Describe the etiopathogenesis of primary congenital glaucoma (PCG) including trabeculodysgenesis
- Recognise the classic clinical triad: epiphora, photophobia, and blepharospasm in an infant
- Identify buphthalmos and Haab striae as physical signs of raised intraocular pressure in the developing eye
- Outline the investigative approach including IOP measurement under anaesthesia and gonioscopy
- Distinguish PCG from secondary causes and from other conditions producing infantile epiphora or corneal clouding
- Describe the surgical management principles (goniotomy and trabeculotomy) and state why surgery is first-line in PCG
INSTRUCTIONS
Congenital glaucoma is a rare but vision-threatening condition in which a developmental defect in the aqueous drainage angle leads to raised intraocular pressure from birth or early infancy. If not recognised and treated promptly, the resulting optic nerve damage causes irreversible blindness in a child who has decades of potential vision ahead. This module uses the OP arc (presentation → anatomy/pathophysiology → examination → diagnosis → management → self-assessment) to build a systematic understanding of the condition. Engage actively with the clinical scenario in the hook and test yourself with the micro-quiz as you progress.
References
- AK Khurana — Comprehensive Ophthalmology, 7th edition, Chapter: Glaucoma (textbook)
- Parsons' Diseases of the Eye, 23rd edition, Chapter: Developmental Anomalies and Congenital Conditions (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 4-month-old boy is brought by his mother who is worried about constant tearing from both eyes, especially in sunlight. She has noticed that when she takes him outdoors, he squints intensely and buries his face in her shoulder. On examination by the paediatrician, both corneas appear larger than expected and slightly hazy. The child cries when the torch is shone in his eyes. The paediatrician refers him urgently to ophthalmology with a suspicion of congenital glaucoma. What is happening in this infant's eyes, and why does acting within weeks — not months — determine whether he will see clearly for the rest of his life?
WHY THIS MATTERS
Primary congenital glaucoma (PCG) affects approximately 1 in 10,000 to 1 in 12,500 live births, making it the commonest form of childhood glaucoma worldwide. Although rare in absolute terms, it is one of the leading preventable causes of childhood blindness globally, and Indian data suggests a higher prevalence in certain communities (Romany/gypsy populations and some consanguineous families). The MBBS graduate must be equipped to recognise the clinical triad (epiphora, photophobia, blepharospasm) and understand that this is a surgical emergency — delay allows progressive optic nerve damage during a critical window when the infant's visual cortex is still plastic. Early surgical success rates exceed 80-90%, making prompt diagnosis life-altering. As a future clinician who will encounter infants in general practice, primary care, or paediatrics, your recognition of the triad triggers the urgent ophthalmology referral that can change the course of a child's life.
RECALL
Before exploring congenital glaucoma, recall your Year 1 anatomy and physiology of the aqueous humour circulation. The aqueous humour is a clear fluid produced continuously by the ciliary body (non-pigmented epithelium) via active secretion and ultrafiltration. It flows from the posterior chamber through the pupil into the anterior chamber and drains primarily through the trabecular meshwork at the iridocorneal angle into Schlemm's canal, then into episcleral veins. A smaller fraction drains via the uveoscleral route. The balance between production and drainage determines intraocular pressure (IOP); the normal range is 10–21 mmHg. Critically, the trabecular meshwork is an embryologically derived structure — any arrest or maldevelopment of the angle will obstruct outflow and elevate IOP. Also recall that the infant sclera and cornea are far more distensible than the adult equivalent, because collagen crosslinks are immature. This is the biological reason why raised IOP in infancy stretches and enlarges the eye rather than simply damaging the optic nerve as it does in adults.
The Triad that Triggers Suspicion
The clinical presentation of primary congenital glaucoma is remarkably specific, driven by the combination of elevated intraocular pressure and the immature, distensible tissues of the infant eye. Rather than the silent, painless field loss that characterises primary open-angle glaucoma in adults, congenital glaucoma makes itself known through three cardinal features that any mother (or a vigilant clinician) can observe: epiphora (excessive tearing), photophobia (light sensitivity), and blepharospasm (involuntary forceful eyelid closure). These symptoms arise because the elevated IOP and corneal stretching irritate the richly innervated corneal epithelium and stroma, generating a pain-and-reflex-closure arc. The photophobia in particular is striking — infants may cry violently when exposed to daylight and remain calm in dimly lit rooms, which parents sometimes misattribute to 'temperament.'
The presentation can be present from birth (true congenital, usually bilateral and often detected in the neonatal period) or may appear in the first few months of life (infantile glaucoma, the commonest subgroup). A minority present between ages 3 and 10 (juvenile congenital glaucoma), in which the globe is less distensible and the presentation is more similar to adult POAG with field loss rather than enlargement.
The corneal haziness that worried the mother in the opening case is a direct consequence of corneal oedema from raised IOP compressing the corneal endothelium, which cannot actively deturgese enough fluid to keep the stroma clear. In early stages the haze may be intermittent; in established disease the cornea becomes permanently cloudy, further reducing visual acuity and providing a physical substrate for amblyopia.
Key clinical features at presentation:
- Epiphora (often mistaken for nasolacrimal duct obstruction)
- Photophobia and blepharospasm (the 'sun-shy' baby)
- Enlarged, hazy cornea (buphthalmos — 'ox eye')
- Corneal diameter >12 mm in a neonate (normal neonatal cornea ≈10 mm)
- Myopia developing from axial elongation of the globe
Anatomy of Failure: Trabeculodysgenesis
The fundamental pathological mechanism in primary congenital glaucoma is trabeculodysgenesis — an arrest or maldevelopment of the trabecular meshwork and anterior chamber angle during embryological development, occurring in the absence of any associated systemic syndrome or ocular anomaly. Understanding this requires appreciation of how the angle forms normally.
During embryogenesis, neural crest cells migrate into the periocular mesenchyme and differentiate to form the corneal stroma, endothelium, and the angle structures. Between the third and seventh month of fetal development, the iridocorneal angle undergoes a progressive maturation process in which the angle tissue 'clefts' away from the cornea and iris root, and the trabecular meshwork acquires its sieve-like architecture. In PCG, this maturation process is incomplete: the angle contains an undifferentiated cellular membrane or a high iris insertion that buries or functionally obstructs the trabecular meshwork. Aqueous outflow is thus mechanically impaired from birth, causing IOP to rise progressively.
The genetic basis is heterogeneous. Mutations in the CYP1B1 gene (encoding cytochrome P450 1B1, expressed in developing angle structures) account for the majority of familial PCG cases, especially in South Asian and Middle Eastern populations. The inheritance is autosomal recessive in most families, which explains the higher prevalence in consanguineous populations. A smaller proportion involves mutations in LTBP2 and MYOC genes.
IMPORTANT DISTINCTION: PCG involves only angle maldevelopment (trabeculodysgenesis). SECONDARY congenital glaucoma, by contrast, occurs when another primary condition — Sturge-Weber syndrome, aniridia, Axenfeld-Rieger syndrome, congenital rubella, or a persistent fetal vasculature — independently damages or obstructs the angle. This distinction matters because secondary causes require treatment of the underlying condition in addition to glaucoma management.
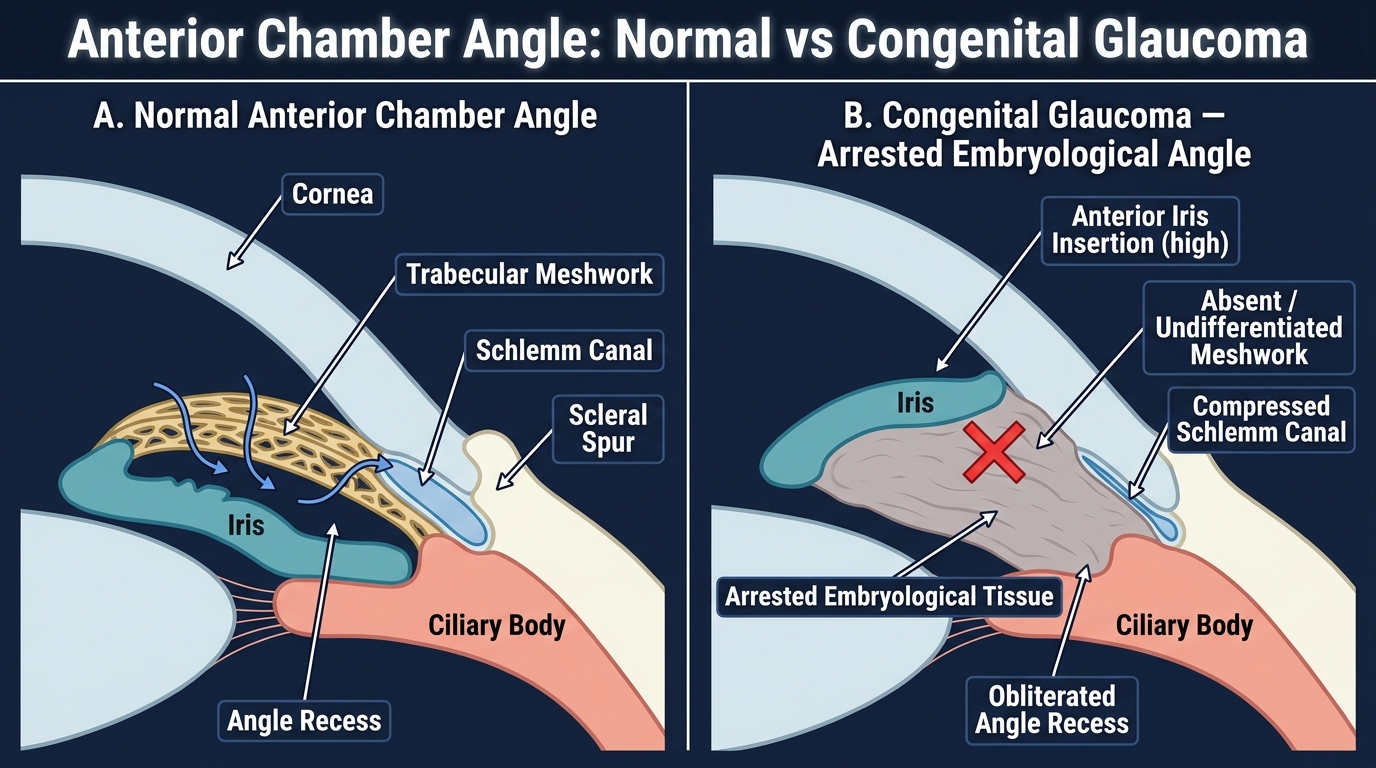
Anterior Chamber Angle: Normal vs Congenital Glaucoma (Arrested Embryological Development)
Examining the Infant: Key Findings
Examining a potentially glaucomatous infant is both clinically and logistically challenging. Infants do not co-operate for slit-lamp examination or IOP measurement, and the diagnostic work-up typically requires an examination under general anaesthesia (EUA). However, several signs can be assessed in the outpatient setting before EUA and form the basis for the referral decision.
On casual inspection, buphthalmos — the enlarged 'ox eye' — is the most visually dramatic sign. The cornea appears larger than normal (diameter >12 mm in a neonate, >13 mm in a child under 2 years) and may appear bluish-grey due to corneal oedema. On careful slit-lamp or loupe examination, Haab striae can be seen: these are horizontal or oblique breaks in Descemet's membrane caused by the physical stretching of the cornea under raised IOP. They run parallel or oblique to the limbus, unlike the vertical striae of forceps delivery trauma, and represent permanent scars that mark previous episodes of corneal stretching. Once formed, Haab striae persist even after IOP is normalised.
Under anaesthesia (EUA), the following are measured and recorded systematically:
- IOP: measured with a Perkins handheld tonometer or pneumotonometer. Note that general anaesthesia (especially halothane) lowers IOP, so any reading >21 mmHg under anaesthesia is very significant. Ketamine anaesthesia, conversely, raises IOP and is avoided.
- Corneal diameter: measured with calipers; a horizontal corneal diameter (white-to-white) >12 mm in a neonate is diagnostic support.
- Gonioscopy: examination of the angle using a Koeppe lens under anaesthesia. This directly visualises the trabeculodysgenesis — the angle is open but shows an absent or dysplastic trabecular meshwork, a high iris insertion, or a granular/anterior uveal tissue overlying the meshwork.
- Optic disc assessment: a cup:disc (C:D) ratio >0.3 in an infant (normal neonatal C:D ≈0.1–0.2) suggests significant glaucomatous damage. Importantly, optic disc cupping in infants can partially REVERSE after surgical IOP reduction, unlike adults — this is a key biological difference.
- Axial length: B-scan biometry confirms globe elongation.
IMPORTANT CLINICAL DISTINCTION: Because infants cannot express corneal pain, Haab striae and corneal oedema serve as surrogate markers for chronic elevated IOP. The parent's history of photophobia and epiphora is the earliest warning.
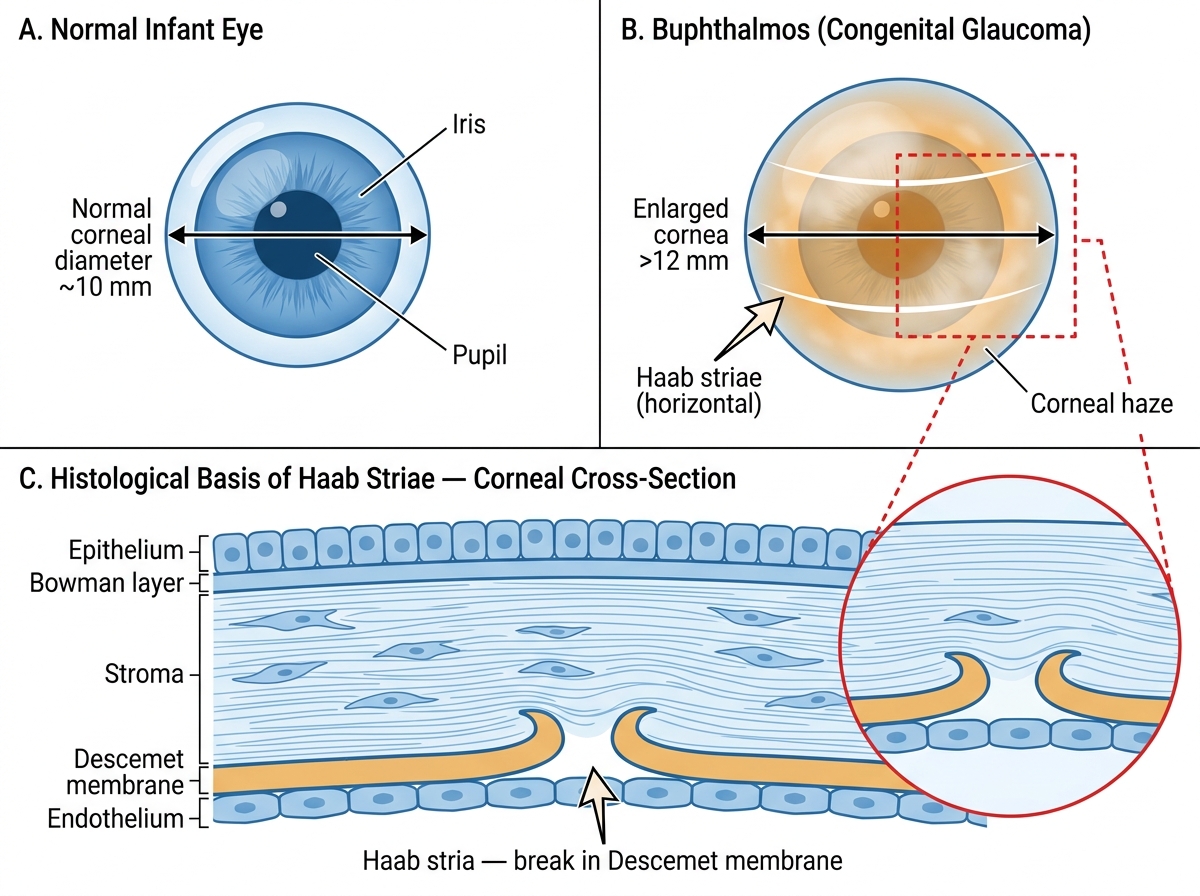
Buphthalmos vs Normal Infant Eye with Haab Striae
SELF-CHECK
A 3-month-old infant presents with epiphora, photophobia, and an enlarged cornea (diameter 13 mm). Under anaesthesia, IOP is 28 mmHg. Gonioscopy shows a high iris insertion with no visible trabecular meshwork. What is the most likely diagnosis?
A. Nasolacrimal duct obstruction
B. Primary congenital glaucoma (trabeculodysgenesis)
C. Peters anomaly (congenital corneal opacity)
D. Anterior uveitis
Reveal Answer
Answer: B. Primary congenital glaucoma (trabeculodysgenesis)
The combination of epiphora + photophobia + blepharospasm (classic triad), an enlarged cornea >12 mm, IOP >21 mmHg under anaesthesia, and gonioscopic demonstration of a high iris insertion with absent trabecular meshwork is the hallmark of primary congenital glaucoma (trabeculodysgenesis). Nasolacrimal duct obstruction causes epiphora but NOT enlarged cornea, raised IOP, or photophobia. Peters anomaly causes corneal opacity but a different gonioscopic picture. Uveitis does not produce this specific angle finding.