Page 3 of 10
OR7.1 | Metabolic Bone Disease Assessment — SDL Guide (Part 3)
Paget's Disease: Clinical Features, Investigations, and Complications
Many patients with Paget's disease are asymptomatic, discovered incidentally on X-ray or a blood test showing a markedly elevated alkaline phosphatase. When symptomatic, the cardinal features relate to the affected bone(s). Bone pain is deep, constant, and worse at rest (unlike mechanical pain) — a distinguishing feature. Deformity is characteristic: bowing of the femur (anterior bowing, producing a sabre-tibia appearance in tibial Paget's) and progressive skull enlargement causing the patient's hat size to increase (an historically classic complaint) and facial bone enlargement (leontiasis ossea, or leonine facies, in severe skull involvement). Skull involvement may compress cranial nerves — sensorineural or mixed hearing loss from cochlear involvement or ossicular chain distortion is the most common cranial nerve complication; optic nerve compression causing visual loss is less common. Spinal cord and nerve-root compression from pagetic vertebrae causes radiculopathy or myelopathy. A highly vascular pagetic skeleton acts as a massive arteriovenous fistula: extensive Paget's disease can generate enough additional cardiac output to cause high-output cardiac failure — a classic but underemphasised complication seen in widespread polyostotic disease, particularly in patients with pre-existing cardiac disease.
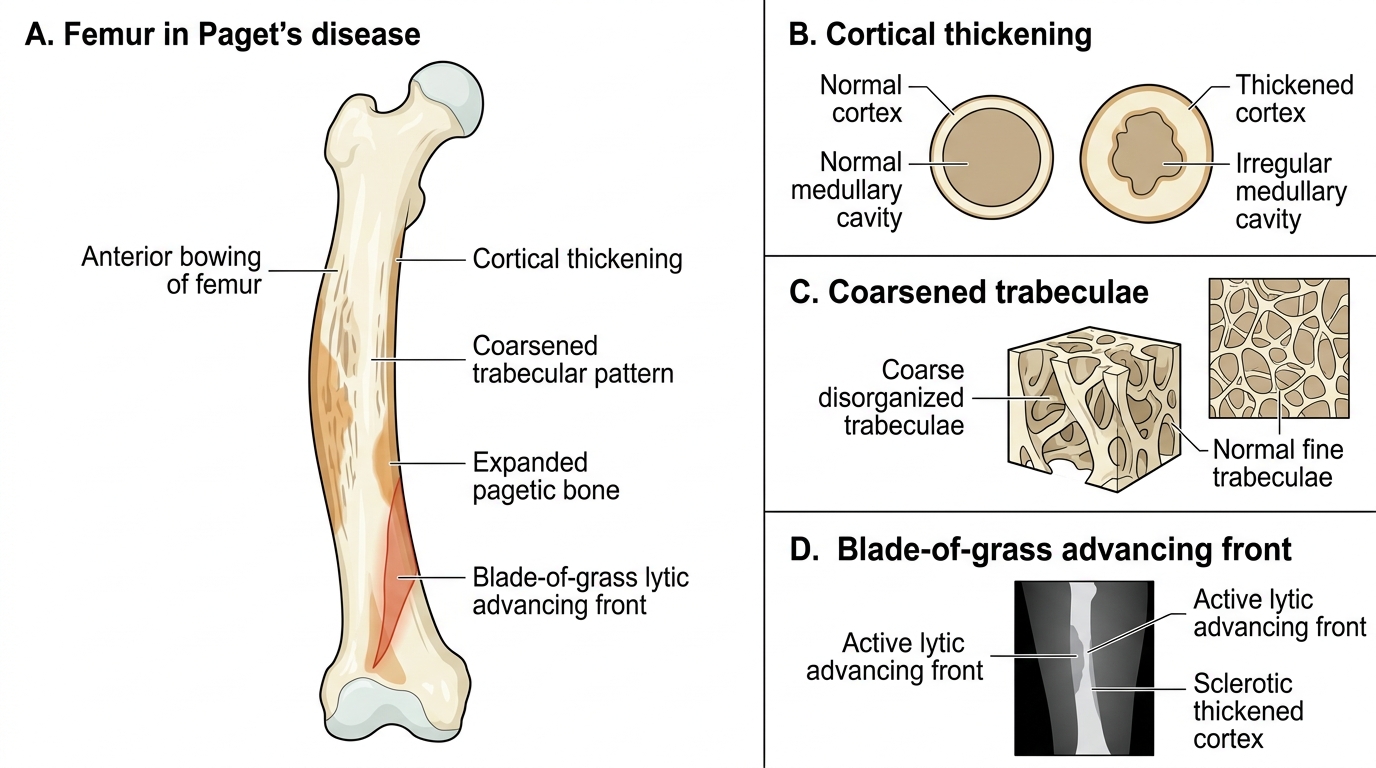
Paget's Disease of the Femur
The biochemical hallmark of Paget's disease is markedly elevated serum alkaline phosphatase (ALP) — often 5-20 times normal — reflecting the intense osteoblastic activity. Serum calcium and phosphate are typically normal in uncomplicated Paget's, distinguishing it clearly from osteomalacia and hyperparathyroidism. PTH is normal. In prolonged immobilisation of a patient with widespread Paget's, however, continued osteoclastic activity can cause hypercalcaemia and hypercalciuria — a clinical trap. Urinary hydroxyproline and serum bone-specific ALP or P1NP (procollagen type I N-terminal propeptide) are sensitive markers of disease activity. Radiological hallmarks include: coarsened trabecular markings, cortical thickening, cotton-wool appearance of the skull (alternating lytic and sclerotic areas producing a heterogeneous mottled pattern), picture-frame vertebra (cortical thickening around the periphery of a vertebra with coarsened internal trabeculae), and the advancing lytic front. Bone scan (technetium-99m methylene diphosphonate scintigraphy) is the most sensitive investigation for identifying all affected skeletal sites and guiding treatment monitoring. A serious but uncommon complication is sarcomatous transformation — pagetic bone occasionally undergoes malignant change to osteosarcoma, fibrosarcoma, or chondrosarcoma; a sudden increase in pain at a pagetic site or rapid rise in ALP should prompt urgent MRI and biopsy.
Biochemical Differentiation: The Metabolic Fingerprint
A fundamental skill in orthopaedic and general medical diagnosis is reading the biochemical panel as a pattern rather than as individual values in isolation. Each of the four metabolic bone diseases disrupts the Ca/PO4/ALP/PTH/vitamin D regulatory circuit at a different point, producing a distinct biochemical signature that is both internally logical and diagnostically specific. The key insight is that these values are not independent numbers to memorise: they are the readout of the bone mineral regulatory system, and when you understand the mechanism of each disease, the biochemical changes become inevitable and predictable rather than arbitrary facts. For example, if you understand that osteomalacia is caused by vitamin D deficiency leading to poor intestinal calcium and phosphate absorption, then low Ca, low PO4, high PTH (compensating), and low vitamin D with high ALP (osteoblasts straining to mineralise bone they cannot mineralise) follows directly from first principles. Similarly, if you understand that Paget's disease is focal disorganised remodelling without systemic mineral depletion, then normal Ca and PO4 with dramatically elevated ALP (osteoblastic hyperactivity) is exactly what you would predict. This section consolidates all four fingerprints side by side so you can practice reading the panel as a diagnostic tool.
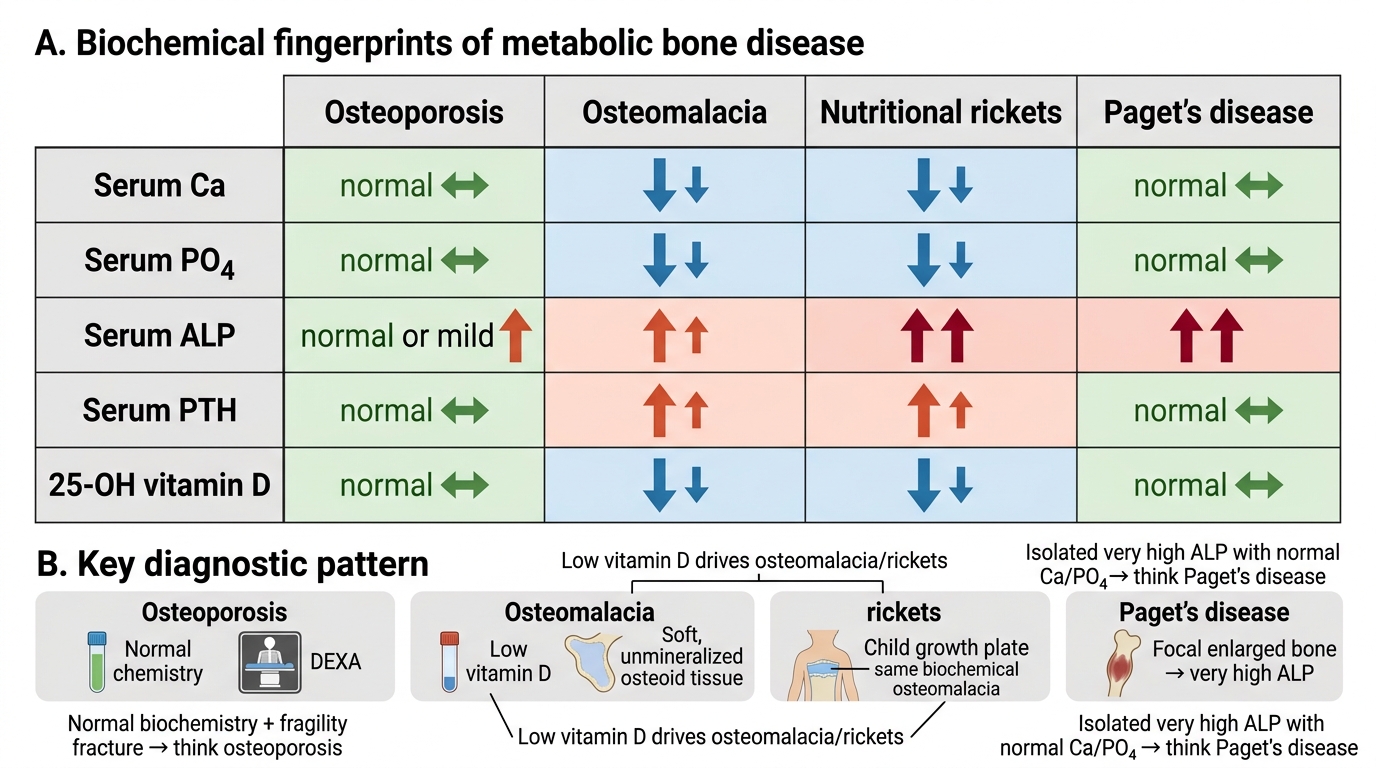
Biochemical Fingerprints of Metabolic Bone Disorders
In osteoporosis: serum calcium is normal (mineral regulation is intact), serum phosphate is normal, ALP is normal or only mildly elevated in severe active disease, PTH is normal, vitamin D is normal. The DEXA T-score is the diagnostic tool, not the biochemistry — a normal biochemical panel in a patient with a fragility fracture should point immediately to osteoporosis.
In osteomalacia (vitamin D deficiency-driven): serum calcium is low (insufficient intestinal absorption), serum phosphate is low (PTH-driven renal wasting plus insufficient absorption), ALP is high (osteoblasts are working overtime trying to deposit osteoid but the mineral is absent), PTH is high (secondary hyperparathyroidism compensating for hypocalcaemia), vitamin D (25-OH) is low.
In nutritional rickets: the biochemical fingerprint is identical to osteomalacia — low Ca, low PO4, high ALP (very high in children — often 3-10 times normal), high PTH, low vitamin D. The difference is clinical and radiological, not biochemical.
In Paget's disease: serum calcium is normal (focal remodelling does not deplete systemic mineral stores under usual conditions), serum phosphate is normal, ALP is very high — the most dramatically elevated of all four conditions, often 5-20 times normal, reflecting intense osteoblastic activity at pagetic sites, PTH is normal, vitamin D is normal.
SELF-CHECK
A 65-year-old man complains that his hat no longer fits and has had deep aching thigh pain for 2 years. His serum calcium is 9.8 mg/dL (normal), phosphate 3.5 mg/dL (normal), ALP 890 U/L (normal <120), PTH normal, vitamin D normal. Skull X-ray shows a mixed lytic and sclerotic pattern. What is the most likely diagnosis and which single investigation would most accurately map all affected skeletal sites?
A. Osteomalacia — 25-OH vitamin D level will confirm
B. Primary hyperparathyroidism — parathyroid hormone assay is diagnostic
C. Paget's disease — technetium-99m bone scintigraphy maps all sites
D. Multiple myeloma — serum protein electrophoresis is diagnostic
Reveal Answer
Answer: C. Paget's disease — technetium-99m bone scintigraphy maps all sites
This is classic Paget's disease: hat size increase (skull enlargement), deep aching bone pain, massively elevated ALP (approximately 7 times normal) with normal calcium, phosphate, PTH, and vitamin D, and the pathognomonic mixed lytic-sclerotic cotton-wool skull appearance. Technetium-99m methylene diphosphonate (MDP) bone scintigraphy is the most sensitive investigation for identifying all affected pagetic sites — areas of intense tracer uptake correspond to pagetic bone activity. Multiple myeloma would show lytic lesions without sclerosis and abnormal protein electrophoresis. Osteomalacia would show low calcium and low phosphate.
Osteomalacia: Treatment
The treatment of nutritional osteomalacia is straightforward and highly effective when given early, before extensive pseudofractures or deformity have occurred. The cornerstone is vitamin D replacement: in adults, ergocalciferol (vitamin D2) or cholecalciferol (vitamin D3) is given in loading doses — typically 60,000 IU weekly for 8-12 weeks, followed by maintenance supplementation of 1000-2000 IU daily. Adequate calcium supplementation (1-1.5 g elemental calcium daily) is essential alongside vitamin D, because as the vitamin D level is restored and intestinal absorption improves, the skeleton's demand for mineralisation can transiently lower serum calcium (the 'hungry bone syndrome' variant of this mechanism). For Looser's zones that have propagated to complete fractures, standard orthopaedic fracture management applies; however, immobilisation should be minimised because osteomalacic bone does not heal well in casts without concurrent metabolic correction. Renal causes (hypophosphataemia) require phosphate supplements and calcitriol rather than ergocalciferol. Monitoring response: ALP falls progressively over weeks to months as unmineralised osteoid is mineralised; a normalising ALP plus resolution of bone pain and muscle weakness confirms adequate treatment. Serum 25-hydroxyvitamin D levels should be checked at 3 months to confirm repletion to >30 ng/mL. Dietary advice (oily fish, egg yolks, fortified dairy) and supervised sun exposure are adjuncts.
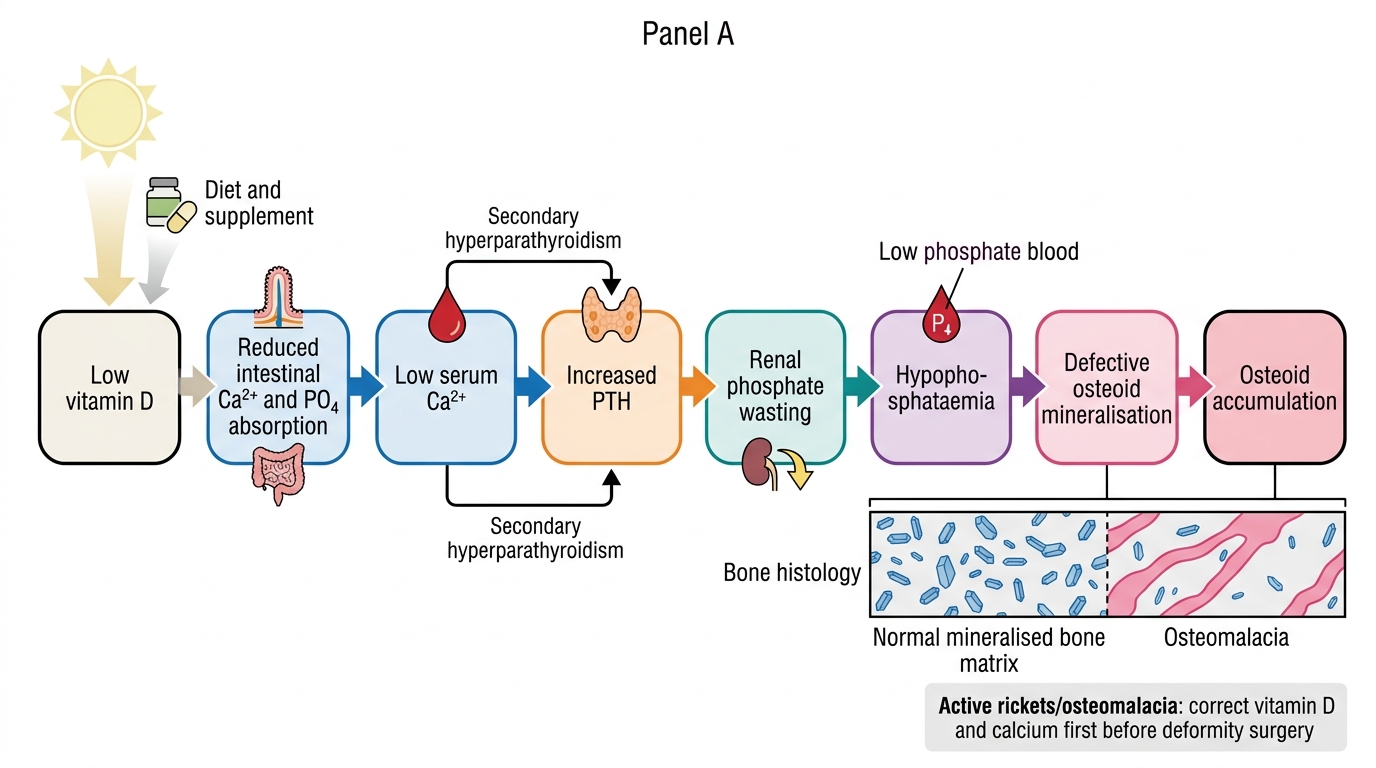
Vitamin D Deficiency Osteomalacia: Pathophysiological Cascade
SELF-CHECK
A 3-year-old child presents with bilateral bow legs (genu varum), widened rachitic wrists, and rachitic rosary. ALP is 650 U/L (normal <300). The mother reports exclusive breastfeeding until 18 months with no vitamin D supplementation. What is the immediate first step in management?
A. Corrective osteotomy of the femur to straighten the bow legs
B. Hemi-epiphysiodesis of the medial distal femoral physis
C. Vitamin D replacement with calcium supplementation and metabolic correction first
D. Referral for full-length leg brace to prevent progression
Reveal Answer
Answer: C. Vitamin D replacement with calcium supplementation and metabolic correction first
No surgical correction should be attempted while rickets is metabolically active. The active disease with elevated ALP (650 U/L) and widened physes must be controlled first with vitamin D and calcium supplementation. The growing skeleton has substantial remodelling capacity — many deformities in children under 2-3 years correct spontaneously with metabolic treatment alone. Osteotomy or hemi-epiphysiodesis is reserved for residual deformity after metabolic control is confirmed (normalised ALP, replete vitamin D). Operating on softened, demineralised bone risks poor fixation, fracture, and deformity recurrence.