Page 4 of 30
PE20.2 | Acute Post Streptococcal Glomerulonephritis — SDL Guide
Learning Objectives
- Enumerate the aetiological agents and nephritogenic strains of Group A Streptococcus responsible for APSGN
- Describe the immune-complex pathogenesis, complement activation pattern, and histological findings in APSGN
- Recognise the clinical features of the nephritic syndrome and differentiate APSGN from other glomerulonephritides
- Plan the appropriate investigation including serological markers, complement studies, and urine analysis
- Outline supportive management and the treatment of complications including hypertensive encephalopathy and acute renal failure
- State the prognosis of APSGN and indications for renal biopsy
INSTRUCTIONS
Acute post-streptococcal glomerulonephritis is the prototype of post-infectious immune-complex nephritis and the commonest cause of acute nephritic syndrome in children in developing countries including India. A thorough understanding of APSGN provides the conceptual template for all other immune-mediated glomerulonephritides and is directly applicable to the ward-level management of a child presenting with haematuria, oedema, and hypertension following a skin or throat infection.
References
- Ghai Essential Pediatrics, 9th edition, Ch 18 — Nephrology (textbook)
- Nelson Textbook of Pediatrics, 21st edition, Ch 527 — Acute Glomerulonephritis (textbook)
- IAP Standard Treatment Guidelines — Acute Glomerulonephritis (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 9-year-old boy presents to the paediatric ward with puffiness of the face for 3 days, decreased urine output, and passage of dark 'cola-coloured' urine. His mother recalls that he had a sore throat 12 days ago, which resolved without antibiotics. On examination he is hypertensive (BP 148/96 mmHg), has periorbital and ankle oedema, and his urine dipstick shows 3+ blood and 2+ protein. His serum creatinine is mildly elevated. How do you explain the connection between his sore throat 12 days ago and his kidneys today? What is the single most likely diagnosis, and what investigations will you order?
WHY THIS MATTERS
Acute post-streptococcal glomerulonephritis (APSGN) is the commonest cause of acute nephritic syndrome in children and the paradigm case through which medical students and clinicians learn the entire class of immune-complex-mediated glomerular diseases. Beyond its own clinical importance, mastering APSGN provides the intellectual scaffolding for understanding lupus nephritis, IgA nephropathy, and membranoproliferative GN — all of which share the language of immune-complex deposition, complement activation, and glomerular inflammation. In India, where Group A streptococcal skin infections (impetigo, pyoderma) remain prevalent in children, APSGN is a genuine public-health concern, and a clinician who misses the diagnosis or mismanages hypertensive encephalopathy puts the patient at risk of stroke, heart failure, or death. This module makes the pathogenesis concrete, the diagnostic workup systematic, and the management actionable.
RECALL
Before proceeding, recall:
- Complement system (from PY/Biochemistry): the alternative pathway is activated by microbial surfaces directly (bypasses C1/C4), leading to C3 cleavage without consuming C4. In contrast, the classical pathway (triggered by antigen-antibody complexes) consumes both C3 AND C4. Knowing this pathway predicts the C3-low/C4-normal pattern of APSGN.
- Hypersensitivity reactions: Type III = immune-complex-mediated (antigen + antibody → complex → complement activation → tissue damage). APSGN is the textbook Type III example in the kidney.
- Nephrotic vs nephritic syndrome distinction: keep these clearly separate — they share the word 'nephro' but differ fundamentally in pathophysiology, clinical features, and management. The nephritic syndrome of APSGN features haematuria, hypertension, and oliguric renal impairment; the nephrotic syndrome (e.g. MCNS) features massive proteinuria, hypoalbuminaemia, oedema, and hyperlipidaemia WITHOUT prominent haematuria or hypertension.
Clinical Presentation of APSGN
APSGN presents most commonly in children aged 5–15 years, with a peak at 6–10 years, and is rare under 3 years. Boys are affected approximately twice as often as girls. The hallmark is the acute nephritic syndrome — a constellation of haematuria, oedema, hypertension, and oliguria that develops after a characteristic latent interval following streptococcal infection. Two infection sites produce two distinct latent intervals: pharyngitis is followed by nephritic symptoms after 1–2 weeks, while impetigo/pyoderma (skin infection) has a longer latent period of 3–6 weeks. This latent period reflects the time required for immune-complex formation and glomerular deposition — it is a clinically crucial history point that distinguishes APSGN from IgA nephropathy (which causes synpharyngitic haematuria with no latent period).
The classic presenting features are:
• Haematuria: the urine appears smoky, tea-coloured, or cola-coloured — the most alarming symptom to parents. Macroscopic haematuria is present in about 30% of cases; the majority have microscopic haematuria only.
• Oedema: predominantly periorbital and facial initially (worse in the morning), then generalising to lower limbs and the entire body. The cause is sodium and water retention from reduced GFR and increased aldosterone activity.
• Hypertension: present in >80% of hospitalised cases. In severe cases, hypertension may cause hypertensive encephalopathy — headache, vomiting, visual disturbance, seizures, and altered consciousness.
• Oliguria and reduced GFR: urine output decreases, and there may be mild azotaemia. Frank acute renal failure requiring dialysis occurs in a minority (<5%).
Complications include pulmonary oedema (from fluid overload + hypertension), hypertensive encephalopathy, and — rarely — acute renal failure. The vast majority (>95%) of children recover completely within weeks to months.
Pathophysiology and Aetiology
APSGN is caused by specific nephritogenic strains of Group A beta-haemolytic Streptococcus (GAS, Streptococcus pyogenes). Not all GAS strains are nephritogenic; the relevant M-protein serotypes differ by infection site — throat infections are predominantly M-types 1, 4, and 12, while skin infections implicate M-types 47, 49, 55, and 57. The key nephritogenic antigens are nephritis-associated plasmin receptor (NAPlr) and streptococcal pyrogenic exotoxin B (SPEB), both of which are thought to initiate the glomerular immune response.
The pathogenetic sequence is an immune-complex-mediated (Type III hypersensitivity) process that unfolds as follows: circulating streptococcal antigens bind specific antibodies produced by the host's immune response during the latent period, forming immune complexes. These complexes deposit in the glomerular mesangium and subepithelial space, triggering activation of the complement cascade via the alternative pathway. The critical downstream effects of complement activation are: recruitment of polymorphonuclear cells, release of proteases and reactive oxygen species, and disruption of the glomerular filtration barrier.
Complement pattern — a key diagnostic discriminator: alternative-pathway activation cleaves C3 abundantly but bypasses C1 and C4. Therefore APSGN produces characteristically LOW serum C3 with NORMAL C4. C3 levels begin to fall at the time of presentation and normalise within 6–8 weeks in typical APSGN. Persistently low C3 beyond 8–12 weeks should raise suspicion for an alternative diagnosis — particularly membranoproliferative glomerulonephritis (MPGN), lupus nephritis, or an inherited complement deficiency.
Histologically (on light microscopy, relevant for biopsy cases): diffuse mesangial and endocapillary proliferation, infiltration by polymorphonuclear cells (exudative GN), and swelling of endothelial cells. Immunofluorescence shows granular ('starry sky') deposits of IgG and C3. Electron microscopy reveals characteristic subepithelial electron-dense deposits (humps) — the pathognomonic feature of APSGN.
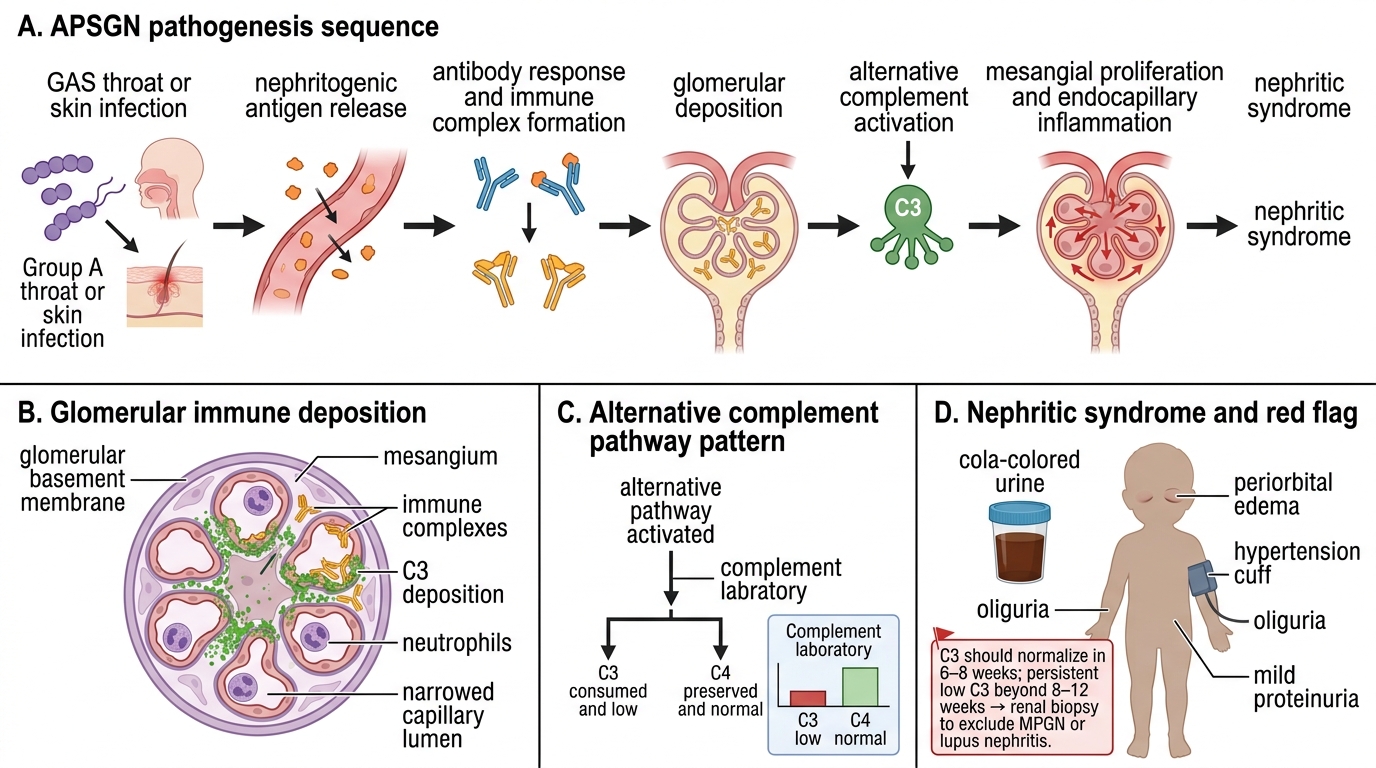
Pathogenesis of Acute Post-Streptococcal Glomerulonephritis
SELF-CHECK
A child with APSGN following throat infection has serum C3 of 30 mg/dL (normal 80–180 mg/dL) and serum C4 of 22 mg/dL (normal 16–47 mg/dL). After 10 weeks, C3 remains at 28 mg/dL. What is the most appropriate next step?
A. Reassure parents; C3 may take up to 6 months to normalise in APSGN
B. Repeat ASO titre to confirm ongoing streptococcal infection
C. Renal biopsy to exclude an alternative cause of persistent hypocomplementaemia
D. Start prednisolone as C3 depression indicates severe disease
Reveal Answer
Answer: C. Renal biopsy to exclude an alternative cause of persistent hypocomplementaemia
In typical APSGN, C3 normalises within 6–8 weeks. Persistent hypocomplementaemia beyond 8–12 weeks is a red flag for an alternative diagnosis — most importantly membranoproliferative GN (MPGN) or lupus nephritis. Renal biopsy is indicated. C4 is NORMAL in APSGN (alternative-pathway activation), so C4 of 22 mg/dL (within normal range) is expected. Reassurance would be inappropriate here; corticosteroids are not indicated for typical APSGN.
Diagnosis and Investigation
Diagnosis of APSGN is clinical in a child with the typical history (preceding streptococcal infection + characteristic latent period) and examination findings (nephritic syndrome), supported by laboratory evidence of streptococcal infection and complement consumption. The key investigations span four categories: urine analysis, blood biochemistry, complement studies, and streptococcal serology.
Understanding the diagnostic logic prevents over-reliance on any single test: culture is usually negative by the time of nephritis (the latent period has elapsed), so serology is the mainstay of microbiological confirmation. The complement pattern tells you which pathway is activated, which in turn guides differential diagnosis. Urine microscopy provides direct glomerular injury evidence — RBC casts are the closest thing to a renal biopsy result you can obtain non-invasively.
Urine analysis:
• Macroscopic haematuria ('cola-coloured' or 'smoky'): 30% of cases. Microscopic haematuria: nearly universal.
• Urine microscopy: dysmorphic red blood cells (acanthocytes, crenated RBCs — deformed during passage through the damaged glomerular filtration barrier) and red cell casts confirm glomerular origin of haematuria.
• Proteinuria: typically subnephrotic (1–2+ on dipstick, <40 mg/m²/hr). Heavy nephrotic-range proteinuria (>40 mg/m²/hr or UPCR >2) is atypical and suggests an overlapping or alternative diagnosis.
Blood biochemistry: serum urea and creatinine (mildly elevated in most; significantly raised in acute renal failure); serum electrolytes (hyperkalaemia if oliguric); FBC (mild anaemia from haemodilution); serum albumin (may be mildly reduced but not as severely as in nephrotic syndrome).
Complement studies:
• Serum C3: LOW — the cornerstone complement finding; present in >90% of APSGN at presentation.
• Serum C4: NORMAL — confirms alternative-pathway activation; a low C4 points towards classical-pathway activation (e.g. lupus nephritis).
• Expected time course: C3 normalises within 6–8 weeks of onset in uncomplicated APSGN.
Streptococcal serology:
• ASO (anti-streptolysin O) titre: elevated in ~75% of post-pharyngitis APSGN; less reliable for post-skin APSGN because streptolysin O is inhibited by skin lipids.
• Anti-DNase B titre: more sensitive for post-impetigo/pyoderma APSGN; should be requested when ASO is normal but clinical suspicion is high.
• Throat culture: usually negative at the time of nephritis (latent period elapsed); useful only if performed during the acute infection.
⚑ AI image — pending faculty review (auto-QA score 4/10; best of 3 attempts)
APSGN vs MCNS: Key Clinical Distinctions
Renal biopsy indications (APSGN is usually diagnosed clinically without biopsy): atypical features warrant biopsy — nephrotic-range proteinuria, absence of streptococcal evidence, C3 persistently low beyond 8–12 weeks, crescentic GN pattern, or failure of clinical improvement within 2–3 weeks.
CLINICAL PEARL
Distinguish APSGN from IgA nephropathy by the latent period. In APSGN, haematuria appears 1–2 weeks after pharyngitis (the time needed for immune-complex formation). In IgA nephropathy, haematuria occurs simultaneously with — or within 24–48 hours of — the pharyngitis (synpharyngitic haematuria — IgA production is immediate). Both present with haematuria after throat infection, but the timing is the key discriminator. Also remember: C3 is LOW in APSGN but NORMAL in IgA nephropathy — check the complement before concluding the diagnosis.