Page 10 of 20
PE21.4 | Systemic Lupus Erythematosus — SDL Guide
Learning Objectives
- Describe the multisystem clinical manifestations of systemic lupus erythematosus in children and adolescents, including the more severe paediatric phenotype compared to adult-onset disease
- Explain the immunopathogenesis of SLE including defective apoptotic clearance, type-I interferon axis, auto-antibody formation, and immune complex-mediated organ injury
- Apply the SLICC 2012 or EULAR/ACR 2019 classification criteria and interpret key serological markers including ANA, anti-dsDNA, anti-Sm, and complement C3/C4
- Describe the ISN/RPS classification of lupus nephritis and its management implications
- Outline the management principles of paediatric SLE including hydroxychloroquine as the anchor drug, glucocorticoids, and immunosuppressive therapy for severe organ involvement
INSTRUCTIONS
Systemic lupus erythematosus is a prototypical autoimmune disease in which the immune system attacks the body's own tissues — affecting virtually every organ system. In children and adolescents, SLE presents earlier, behaves more aggressively, and carries higher rates of nephritis, CNS involvement, and haematological complications than adult-onset disease. The diagnosis requires pattern recognition across multiple organ systems and a working knowledge of serological markers that can appear and disappear with disease activity. Management has improved dramatically, but lupus nephritis — the single most important determinant of long-term outcome — still causes renal failure in 10–15% of paediatric patients. Understanding this disease is essential for any physician who will care for adolescents.
References
- Ghai Essential Pediatrics, 9th Ed, Ch 17 — Systemic Lupus Erythematosus (textbook)
- Nelson Textbook of Pediatrics, 21st Ed, Ch 181 — Systemic Lupus Erythematosus (textbook)
- SLICC Classification Criteria for SLE, 2012 — Petri et al., Arthritis & Rheumatism (guideline)
- EULAR/ACR Classification Criteria for SLE, 2019 (guideline)
- EULAR Recommendations for Management of Paediatric SLE, 2023 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 14-year-old girl presents to the outpatient department with a 3-month history of joint pain (both wrists and fingers), fatigue, and a facial rash that worsens in sunlight. Over the past 2 weeks she has developed swelling of both feet. On examination she has a butterfly-shaped erythematous rash over her cheeks and nasal bridge (sparing the nasolabial folds), tender swelling of the proximal interphalangeal joints bilaterally, and 3+ protein on urine dipstick. Her blood pressure is 138/88 mmHg. CBC shows Hb 8.4 g/dL, WBC 3,100/µL (lymphocytes 600/µL), platelets 92,000/µL. Her ANA returns as 1:1280. Her parents are frightened. You recognise that she likely has SLE with multi-organ involvement — and that the nephrotic proteinuria and thrombocytopenia mean she needs urgent investigation and treatment today, not at a follow-up appointment next week.
WHY THIS MATTERS
Systemic lupus erythematosus (SLE) is one of the most important systemic autoimmune diseases in clinical medicine, affecting approximately 1.5–7.5 per 100,000 children worldwide, with higher prevalence in South Asian, Afro-Caribbean, and East Asian populations. In India, paediatric SLE is underrecognised — the butterfly rash is attributed to 'sunburn,' the joint pain to growing pains, and the anaemia to nutritional deficiency. Paediatric SLE (onset <18 years) differs critically from adult-onset SLE: it is more severe at presentation, has higher rates of lupus nephritis (occurring in 50–80% of children vs ~40% of adults), and carries greater risks of CNS lupus and haematological crises. The stakes are high: without treatment, lupus nephritis progresses to end-stage renal disease; neuropsychiatric lupus causes seizures, stroke, and psychosis; and SLE-related infections and corticosteroid toxicity are major sources of morbidity.
RECALL
Before proceeding, activate your prior knowledge. Recall from Physiology: type III hypersensitivity — immune complex-mediated tissue damage (SLE is the prototypical type III hypersensitivity disease). Recall complement pathways: the classical pathway is activated by antigen-antibody complexes (IgG/IgM), generating C3a/C5a and consuming serum C3 and C4. Recall from Pathology: apoptosis and the clearance of apoptotic bodies — when this fails, intracellular antigens (nuclear DNA, histones, Sm ribonucleoproteins) become exposed to the immune system. Recall from Anatomy: the glomerular structure — mesangium, basement membrane, epithelial (podocyte) and endothelial layers — and how immune complex deposition at different layers produces different patterns of nephritis. These concepts are the structural foundation of SLE pathogenesis.
Clinical Presentation of Paediatric SLE
SLE is the great mimicker — it can involve virtually every organ system, and its presentation can resemble dozens of other conditions. In paediatric practice, the diagnosis is often delayed by six months or more because the early features of fatigue, arthralgia, and anaemia are attributed to far more common and benign conditions such as viral illness, growing pains, or nutritional deficiency. The key to timely diagnosis is recognising the multisystem pattern: when a teenage girl has joint pain AND a facial rash AND anaemia AND low platelets AND haematuria, no single one of these features alone is diagnostic — but together they paint an unmistakable picture of immune-mediated multi-organ inflammation. The SLICC 2012 criteria formalise this pattern recognition by requiring involvement across multiple domains before classifying the disease. Clinical features span eight major organ systems and evolve over months to years.
Constitutional symptoms are nearly universal: prolonged fatigue, fever (low-grade to high, often the first feature and may mimic infection), weight loss, and generalised lymphadenopathy. These are often present for months before the diagnosis is made.
Mucocutaneous features:
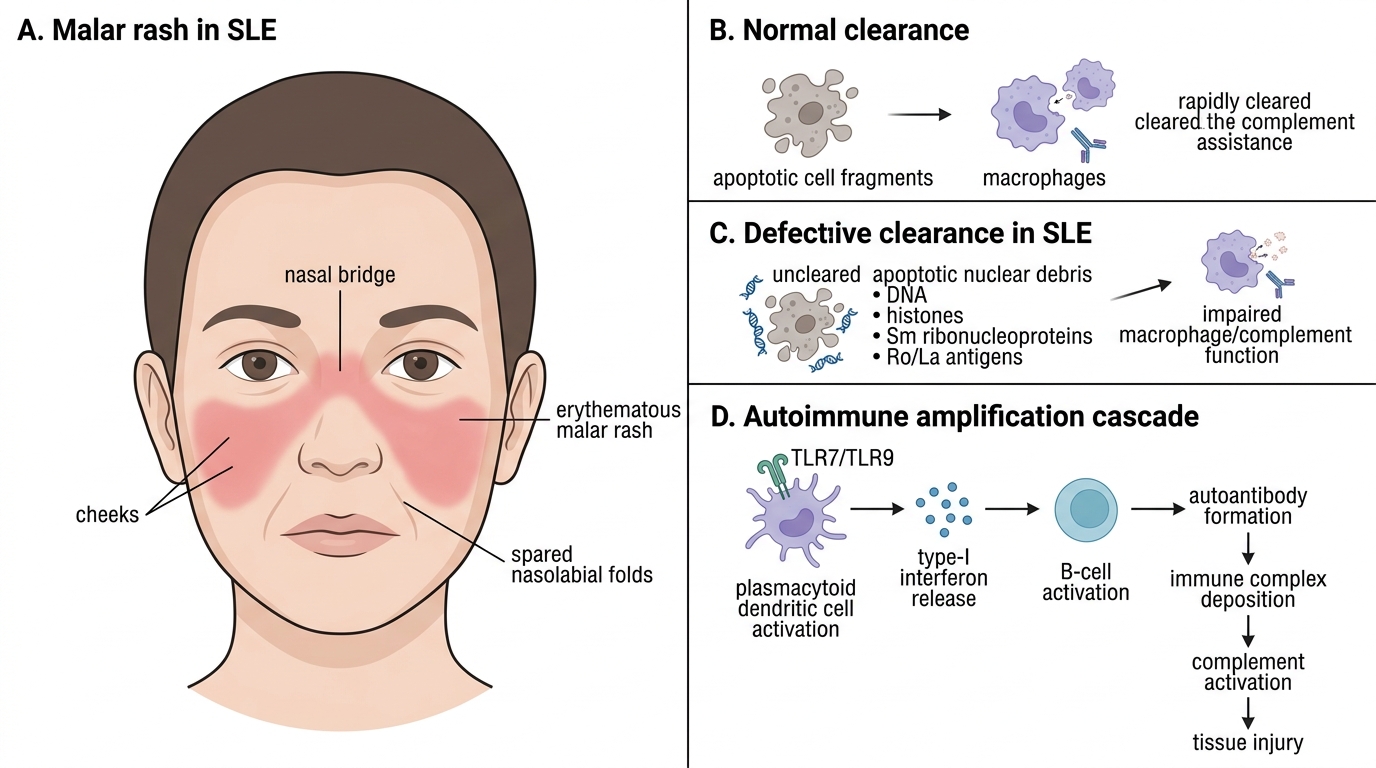
• Malar (butterfly) rash: erythematous, flat-to-raised rash over both cheeks and nasal bridge, characteristically sparing the nasolabial folds — this anatomical sparing is the diagnostic clue distinguishing it from rosacea and seborrhoeic dermatitis, which do involve the folds. It is photosensitive and worsens after sun exposure.
• Discoid rash: chronic scarring plaques with follicular plugging and central atrophy — leaves permanent scars and is less common in children than adults.
• Photosensitivity: exaggerated skin reaction to UV light, including rash or systemic flares after sun exposure.
• Oral ulcers: painless oral or nasopharyngeal ulcers, typically on the hard palate — often discovered on examination, not reported by the patient.
• Non-scarring alopecia: diffuse hair loss, especially frontal 'lupus hair' (thin, fragile, broken hairs along the hairline).
• Raynaud's phenomenon: episodic digital colour changes (white-blue-red) with cold exposure.
Musculoskeletal features: arthritis or arthralgia in 90% of children at some point; typically symmetrical, involving small joints of the hands; non-erosive (unlike RA) — joints return to normal appearance after treatment.
Renal involvement (lupus nephritis): the most important determinant of long-term outcome. Present in 50–80% of children with SLE (higher than adults). Clinical features: haematuria, proteinuria, hypertension, oedema. Nephrotic syndrome, haematuria, and hypertension in an adolescent girl with a facial rash is lupus nephritis until proven otherwise.
Haematological features: normocytic normochromic anaemia (anaemia of chronic disease), haemolytic anaemia (Coombs-positive, immune-mediated red cell destruction), leukopenia (WBC <4,000, especially lymphopenia — lymphocytes <1,000, a SLICC criterion), and thrombocytopenia (<100,000 — immune-mediated platelet destruction by anti-platelet antibodies). Pancytopenia in a teenage girl should always prompt SLE screening.
CNS lupus (NPSLE): seizures, psychosis, cognitive impairment, headaches, cerebrovascular events, peripheral neuropathy. Present in 20–30% of children with SLE at some point — higher than in adult SLE.
Serositis: pleuritis (pleuritic chest pain, pleural rub, effusion) and pericarditis (pericardial friction rub, chest pain, effusion). May present as unexplained polyserositis.
Paediatric-specific severity pattern: compared to adult-onset SLE, paediatric SLE has: higher rates of lupus nephritis, haematological involvement, and CNS lupus; more aggressive serological activity (higher anti-dsDNA, lower complement at diagnosis); more frequent hospitalisations; and greater long-term renal damage.
Malar Rash and Immune Pathogenesis of SLE
Pathophysiology and Aetiology of SLE
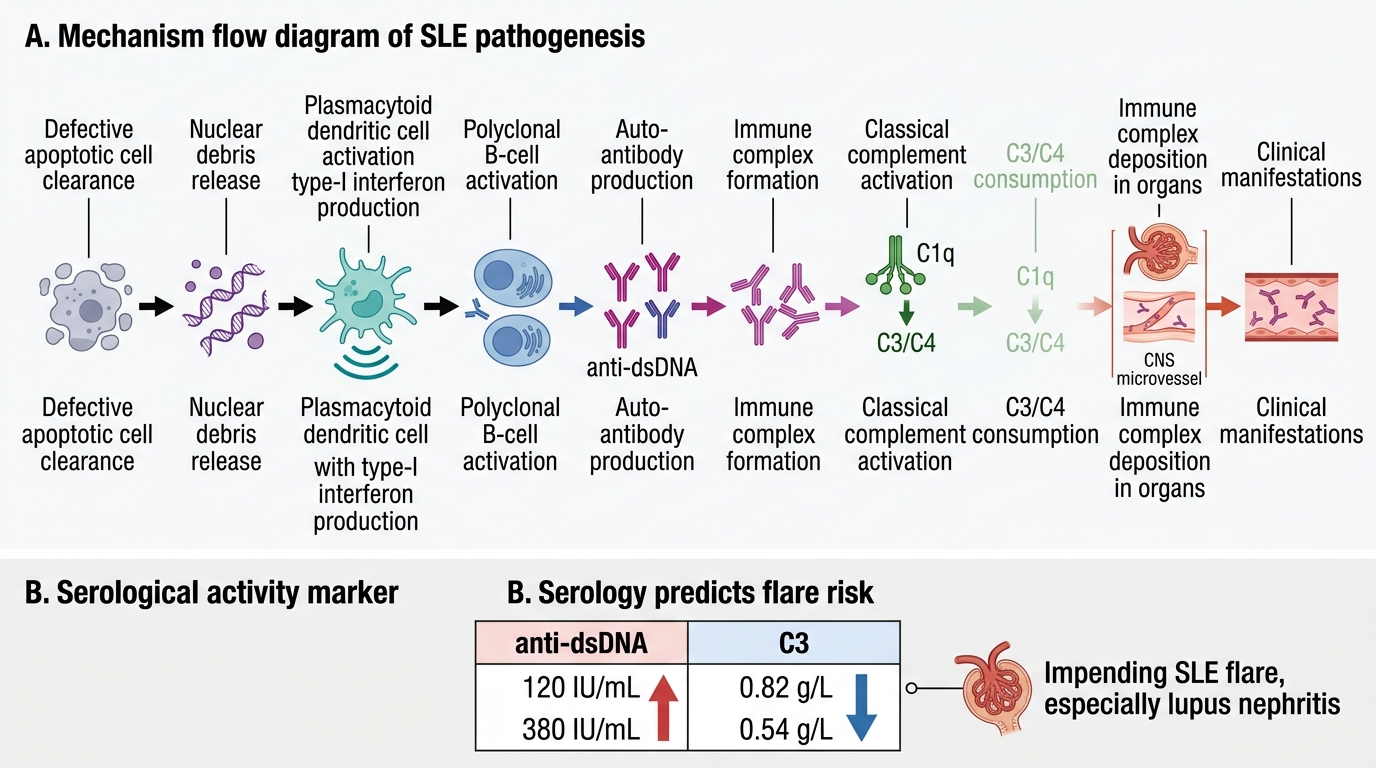
SLE results from a fundamental failure of immune self-tolerance — the loss of the normal mechanisms that prevent the immune system from attacking the body's own nuclear antigens. This failure operates through a cascade that begins with defective clearance of cellular debris and amplifies into a self-sustaining cycle of auto-antibody production, immune complex deposition, and complement-mediated tissue injury.
Step 1 — Defective apoptotic debris clearance: In healthy individuals, when cells undergo apoptosis, nuclear contents (DNA, histones, Sm ribonucleoproteins, Ro/La antigens) are packaged into apoptotic bodies and rapidly cleared by macrophages via complement receptor-mediated phagocytosis. In SLE-susceptible individuals, this clearance is impaired — due to genetic deficiencies of C1q, DNase-I, or complement receptor 1, or due to aberrant macrophage function. Uncleared nuclear debris accumulates and is processed by dendritic cells.
Step 2 — Type-I interferon axis activation: Uncleared nuclear DNA and RNA-containing complexes activate Toll-like receptors (TLR7 and TLR9) on plasmacytoid dendritic cells (pDCs), triggering massive production of type-I interferons (IFN-α/β). The resulting 'interferon signature' (elevated expression of interferon-stimulated genes) is detectable in 75% of SLE patients and correlates with disease activity. Type-I interferons activate B cells, T cells, and dendritic cells, breaking peripheral tolerance.
Step 3 — Polyclonal B-cell activation and auto-antibody production: Activated autoreactive B cells, driven by T-helper cell signals and type-I interferon, produce auto-antibodies against nuclear antigens — including anti-dsDNA, anti-Sm (Smith antigen), anti-histone, anti-Ro/SSA, anti-La/SSB, and antiphospholipid antibodies. These auto-antibodies bind their target antigens, forming immune complexes.
Step 4 — Immune complex deposition and complement consumption: Immune complexes deposit in the glomerular mesangium and subendothelial/subepithelial spaces (causing nephritis), in dermal vessels (causing the rash), in serosal surfaces (causing serositis), and in the choroid plexus (contributing to CNS lupus). Deposited complexes activate the classical complement pathway (IgG/IgM + C1q → C3/C4 cleavage → anaphylatoxins C3a/C5a → neutrophil recruitment → tissue injury). This explains why serum C3 and C4 are low in active SLE — they are being consumed at sites of immune complex deposition.
Genetic and hormonal factors: SLE is strongly influenced by genetic background — HLA-DR2/DR3 associations, C4A null allele, IRF5 polymorphisms. The striking female predominance (9:1 in adults; 4:1 in children) reflects oestrogen-mediated enhancement of B-cell activation and survival. In paediatric SLE, the sex ratio is less extreme before puberty, equalising to approximately 3–4:1.
SLE Pathogenesis and Serological Flare Markers
SELF-CHECK
A 15-year-old girl with known SLE presents for follow-up. Her last visit 3 months ago showed anti-dsDNA titre of 120 IU/mL and C3 of 0.82 g/L. Today her anti-dsDNA is 380 IU/mL and C3 is 0.54 g/L. She has no new symptoms. What do these serological changes indicate?
A. Remission — antibody titres are rising as the immune system recovers
B. An impending flare — rising anti-dsDNA and falling complement predict increased disease activity
C. A drug side effect from hydroxychloroquine — reduce the dose
D. Normal fluctuation — no action needed until symptoms develop
Reveal Answer
Answer: B. An impending flare — rising anti-dsDNA and falling complement predict increased disease activity
Rising anti-dsDNA titre and falling serum complement (C3) are the most reliable serological predictors of an SLE flare, particularly lupus nephritis. Anti-dsDNA correlates directly with disease activity and particularly with nephritis activity — rising titres indicate increased immune complex formation with nuclear antigens. Falling C3 reflects increased complement consumption at sites of immune complex deposition. This serological pattern should prompt clinical re-evaluation (urinalysis, BP, FBC) and may warrant intensification of immunosuppression even before overt symptoms develop. Waiting for symptoms means waiting for organ damage to have already occurred.
Diagnosis and Classification Criteria
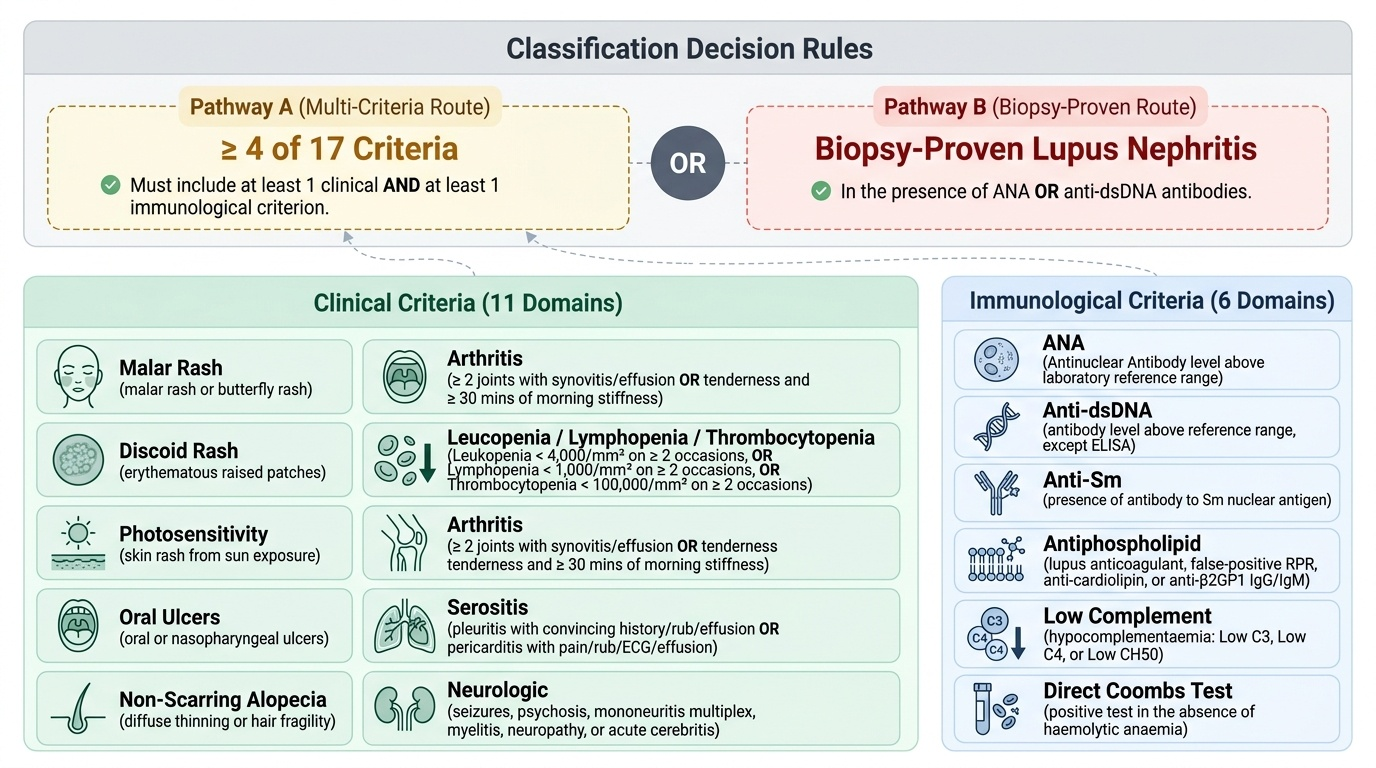
The diagnosis of SLE is clinical, supported by laboratory and immunological data. Two validated classification criteria systems are in current use — the SLICC 2012 criteria and the EULAR/ACR 2019 criteria — both of which were designed for classification in research but are routinely applied in clinical practice. The SLICC 2012 system is more sensitive than the older ACR 1997 criteria, particularly for paediatric SLE, and is the system most widely used in Indian paediatric rheumatology training.
Provided image
SLICC 2012 Classification Criteria: A patient is classified as having SLE if they satisfy ≥4 of 17 criteria (with at least 1 clinical AND at least 1 immunological criterion), OR if they have biopsy-proven lupus nephritis plus a positive ANA or anti-dsDNA. The 11 clinical criteria are: malar rash, discoid rash, oral/nasopharyngeal ulcers, non-scarring alopecia, arthritis (≥2 joints with synovitis), serositis (pleuritis or pericarditis), renal (proteinuria >500 mg/24h or RBC casts), neurological (seizures, psychosis, mononeuritis multiplex, myelitis, peripheral/cranial neuropathy, cerebritis), haemolytic anaemia, leukopenia (<4,000 on ≥2 occasions) OR lymphopenia (<1,000 on ≥2 occasions), thrombocytopenia (<100,000 on ≥2 occasions). The 6 immunological criteria are: ANA above laboratory reference range, anti-dsDNA above reference, anti-Sm, antiphospholipid antibodies, hypocomplementaemia (low C3 OR C4 OR CH50), direct Coombs test positive (without haemolytic anaemia).
EULAR/ACR 2019 Criteria: Entry criterion is ANA positivity ≥1:80 on HEp-2 cells; if negative, SLE classification is excluded. Positive entry criterion leads to weighted scoring across 7 domains (constitutional, haematological, neuropsychiatric, mucocutaneous, serosal, musculoskeletal, renal) plus immunological criteria. A score ≥10 points classifies SLE. This system has higher specificity than SLICC 2012.
Key auto-antibodies and their clinical significance:
• ANA: Present in >95% of SLE patients — the best screening test, but low specificity (also positive in JIA, thyroid disease, Sjögren's, viral infections, normal individuals at low titres). A negative ANA makes SLE very unlikely.
• Anti-dsDNA: Highly specific for SLE (~70% sensitivity, >95% specificity). Titres correlate with disease activity, particularly nephritis — rising anti-dsDNA + falling complement = flare signal.
• Anti-Sm: Highly specific for SLE (~25–30% sensitivity, >99% specificity). Does NOT correlate with disease activity — its presence is diagnostically meaningful but not a monitoring tool.
• Anti-Ro/SSA and anti-La/SSB: Associated with neonatal lupus (passively transferred to the fetus → congenital heart block), Sjögren's overlap, photosensitivity, subacute cutaneous lupus.
• Antiphospholipid antibodies (aPL: anticardiolipin, anti-β2-glycoprotein-I, lupus anticoagulant): Associated with thrombosis (venous and arterial), recurrent pregnancy loss, thrombocytopenia — antiphospholipid syndrome (APS).
Complement C3 and C4: Low C3 and C4 indicate active complement consumption by immune complexes — correlate with nephritis activity. Always check C3 and C4 alongside anti-dsDNA as a flare-monitoring pair.
Lupus nephritis — ISN/RPS Classification (2003, revised 2018):
• Class I: Minimal mesangial — normal light microscopy, mesangial deposits on immunofluorescence; no specific treatment
• Class II: Mesangial proliferative — mesangial cell proliferation; usually mild; hydroxychloroquine + low-dose steroids
• Class III: Focal (<50% glomeruli affected) — immune complex deposits; requires induction immunosuppression
• Class IV: Diffuse (≥50% glomeruli) — the most severe and most common class in paediatric SLE; segmental or global; requires aggressive induction
• Class V: Membranous — subepithelial deposits; nephrotic syndrome is dominant feature; management differs from III/IV
• Class VI: Advanced sclerosing (≥90% sclerosis) — represents end-stage fibrosis; immunosuppression not beneficial
Renal biopsy is indicated in any child with SLE and: persistent proteinuria (>500 mg/24h or protein:creatinine >0.5), haematuria + proteinuria, rising creatinine, nephrotic syndrome, or hypertension — to determine the class and guide treatment intensity.
CLINICAL PEARL
The most reliable flare-monitoring pair in SLE is anti-dsDNA titre rising + complement C3/C4 falling — this combination predicts nephritis flares days to weeks before clinical symptoms or urine changes. Every child with SLE should have anti-dsDNA and complement measured at each clinic visit. When both are moving in the wrong direction, act before the urine dipstick becomes positive. Conversely, anti-Sm does NOT correlate with activity — do not use it to monitor disease. Anti-Sm confirms diagnosis; anti-dsDNA monitors disease.