Page 11 of 34
PE26.4 | Hemolytic Anaemia — SDL Guide (Part 2)
Diagnosis: Confirming Haemolysis and Identifying the Cause
The diagnostic approach to haemolytic anaemia proceeds in two stages. The first stage is confirming that haemolysis is occurring — this uses tests that detect the consequences of accelerated red cell destruction. The second stage is identifying the cause of haemolysis — this uses condition-specific tests targeted at the most likely diagnosis based on the clinical presentation, age, ethnicity, triggering event, and peripheral smear findings. Attempting to run all possible haemolytic workup tests simultaneously without first characterising the smear and clinical picture is inefficient and exposes the patient to unnecessary cost and phlebotomy. The peripheral blood smear is the pivotal bridge between the two stages: it simultaneously confirms haemolytic morphology and points directly at the most likely condition, allowing targeted second-stage testing rather than a broad panel.
Stage 1 — Confirming haemolysis (generic markers):
• Reticulocyte count >2% (or elevated absolute reticulocyte count): indicates a regenerative marrow response to RBC loss; the key differentiator from hypoproliferative normocytic anaemia
• Unconjugated (indirect) bilirubin elevated: from catabolism of haem from destroyed RBCs
• LDH (lactate dehydrogenase) elevated: released from lysed RBCs; also elevated in megaloblastic anaemia (intramedullary haemolysis) and myocardial infarction — not haemolysis-specific but sensitive
• Serum haptoglobin reduced or absent: haptoglobin binds free haemoglobin released from lysed RBCs and is then cleared by the liver; depletion indicates active haemolysis (most useful in intravascular haemolysis)
• Peripheral blood smear: morphological assessment — the single most informative test after the CBC; identifies spherocytes (HS, AIHA), sickle cells, target cells, bite cells, Heinz bodies (supravital stain), fragmented RBCs (schistocytes in MAHA)
| Marker | Haemolysis | Normal |
|---|---|---|
| Reticulocytes | >2% (↑) | <2% |
| Unconjugated bilirubin | Elevated | <1 mg/dL |
| LDH | Elevated | Normal |
| Haptoglobin | ↓ or absent | Normal |
| Urine for Hb | Present (intravascular) | Absent |
Stage 2 — Identifying the cause (condition-specific tests):
• G6PD deficiency: G6PD enzyme assay (fluorescent spot test or quantitative) — perform after the crisis has resolved (falsely high during crisis due to younger cells)
• Thalassaemia major: haemoglobin electrophoresis (HPLC method preferred) — shows HbF markedly elevated, HbA absent or trace; serum ferritin (iron overload assessment)
• Sickle cell disease: haemoglobin electrophoresis (HbS predominant band); sickling test (Na-metabisulphite screen — cheap and rapid)
• Hereditary spherocytosis: osmotic fragility test (spherocytes lyse in 0.5% NaCl vs normal 0.45% NaCl); EMA binding test (flow cytometry — most sensitive)
• AIHA: Direct Coombs test (DAT) — positive; followed by antibody characterisation (IgG vs IgM, thermal amplitude)
• If cause still unclear: haematology referral; consider bone marrow examination to assess erythroid hyperplasia; hereditary haemolytic panel by molecular testing
CLINICAL PEARL
G6PD enzyme assay must be performed AFTER the haemolytic crisis resolves, not during the acute phase. During an acute episode, the most severely G6PD-deficient older cells are selectively destroyed, leaving predominantly younger reticulocytes — which have relatively higher G6PD activity even in G6PD-deficient individuals. Assaying during the crisis may give a falsely 'borderline' or 'normal' result. Wait 2–3 months after the crisis before performing the quantitative G6PD assay.
Management of Haemolytic Anaemias
Management of haemolytic anaemias is highly condition-specific, reflecting the different mechanisms and natural histories of each disorder. There is no single universal treatment; the goal is to maintain adequate haemoglobin to allow normal growth and development, prevent organ damage from anaemia and iron overload, and manage acute crises promptly. The five major conditions covered in this SDL each have distinct management protocols that must be known precisely, because errors in management — particularly in iron chelation timing for thalassaemia, splenectomy timing for HS, and hydroxyurea use in SCD — directly affect long-term outcomes. A common and dangerous error is prescribing supplemental iron for a child with haemolytic anaemia simply because their Hb is low — iron is contraindicated in thalassaemia major and sickle cell disease, where iron overload is already a major complication; iron supplementation is only appropriate in documented concurrent IDA.
Thalassaemia Major — transfusion and chelation:
• Regular transfusion: packed RBCs every 3–4 weeks; target pre-transfusion Hb ≥9–10 g/dL (suppresses ineffective erythropoiesis, prevents bony changes, allows normal growth)
• Iron chelation: transfusion iron loading eventually causes organ damage (liver cirrhosis, cardiac iron overload, endocrine failure — diabetes, hypogonadism); chelation is started when serum ferritin >1000 µg/L, typically after 10–15 transfusions
- Desferrioxamine (DFO): subcutaneous infusion 8–12 hours/night, 5–7 nights/week; very effective but cumbersome
- Deferasirox (oral, once daily): preferred in current practice for compliance
- Deferiprone (oral): most effective at removing cardiac iron
• Splenectomy: for hypersplenism (falling transfusion intervals or rising requirement); after age 5 with pre-splenectomy vaccines + lifelong penicillin prophylaxis post-op
• Bone marrow transplantation (BMT): the only curative option; best outcomes in young, well-chelated patients with an HLA-matched sibling donor; not universally available
G6PD Deficiency — trigger avoidance and supportive care:
• No specific treatment is needed between episodes; the G6PD deficiency itself is not correctable
• During acute haemolytic crisis: stop the offending drug/exposure; provide IV hydration; transfuse if Hb drops severely (Hb <7 g/dL with compromise)
• Patient and family education is the mainstay of management: provide a written list of drugs and foods to avoid (primaquine, dapsone, nitrofurantoin, sulphonamides, fava beans, naphthalene); ensure all future treating clinicians are informed; carry a card
• Screen all male siblings and maternal uncles (X-linked carrier state)
Hereditary Spherocytosis — splenectomy:
• Most children with HS have mild-to-moderate haemolysis that does not require transfusion; monitor Hb and reticulocytes
• Splenectomy is the definitive treatment — removal of the site of spherocyte destruction; haemoglobin normalises, jaundice resolves, gallstone formation stops
• Timing: defer until age 5–6 years minimum to reduce the risk of overwhelming post-splenectomy infection (OPSI) — young children are most vulnerable to encapsulated organisms
• Pre-splenectomy vaccination mandatory: polyvalent pneumococcal vaccine, meningococcal conjugate vaccine, Hib vaccine — at least 2 weeks before surgery
• Post-splenectomy penicillin prophylaxis (oral penicillin V) for at least 5 years after splenectomy, or for life if the patient is immunocompromised
• Folic acid 1 mg/day to meet high folate demand from chronic haemolysis
Sickle Cell Disease — hydroxyurea and prophylaxis:
• Hydroxyurea (hydroxycarbamide): increases HbF production (HbF does not sickle); reduces vasoocclusive crisis frequency by ~50%, reduces acute chest syndrome, reduces transfusion requirement; dose: 15–20 mg/kg/day orally; monitor FBC for myelosuppression
• Penicillin prophylaxis from diagnosis (all children with SCD have functional asplenia by age 5); oral penicillin V twice daily; continues until at least age 5
• Vaccinations: same as post-splenectomy — pneumococcal, meningococcal, Hib; also annual influenza
• Folic acid supplementation (1 mg/day) for chronic haemolysis
• Acute vasoocclusive crisis management: hydration, analgesia (stepwise — paracetamol, NSAIDs, opioids for severe pain), oxygen if hypoxic; blood transfusion for aplastic crisis, splenic sequestration, acute chest syndrome with severe hypoxia
AIHA — immunosuppression:
• Warm AIHA (IgG-mediated, most common in children): oral prednisolone 1–2 mg/kg/day; response expected within 1–3 weeks; taper slowly over 4–6 weeks after response; avoid splenectomy in young children (high failure rate and OPSI risk)
• Refractory cases: rituximab (anti-CD20, B-cell depletion), azathioprine, or ciclosporin
• Transfusion in AIHA requires cross-match with the antibody identified on Coombs test — can be technically challenging; 'most-compatible' blood is used when cross-match is difficult
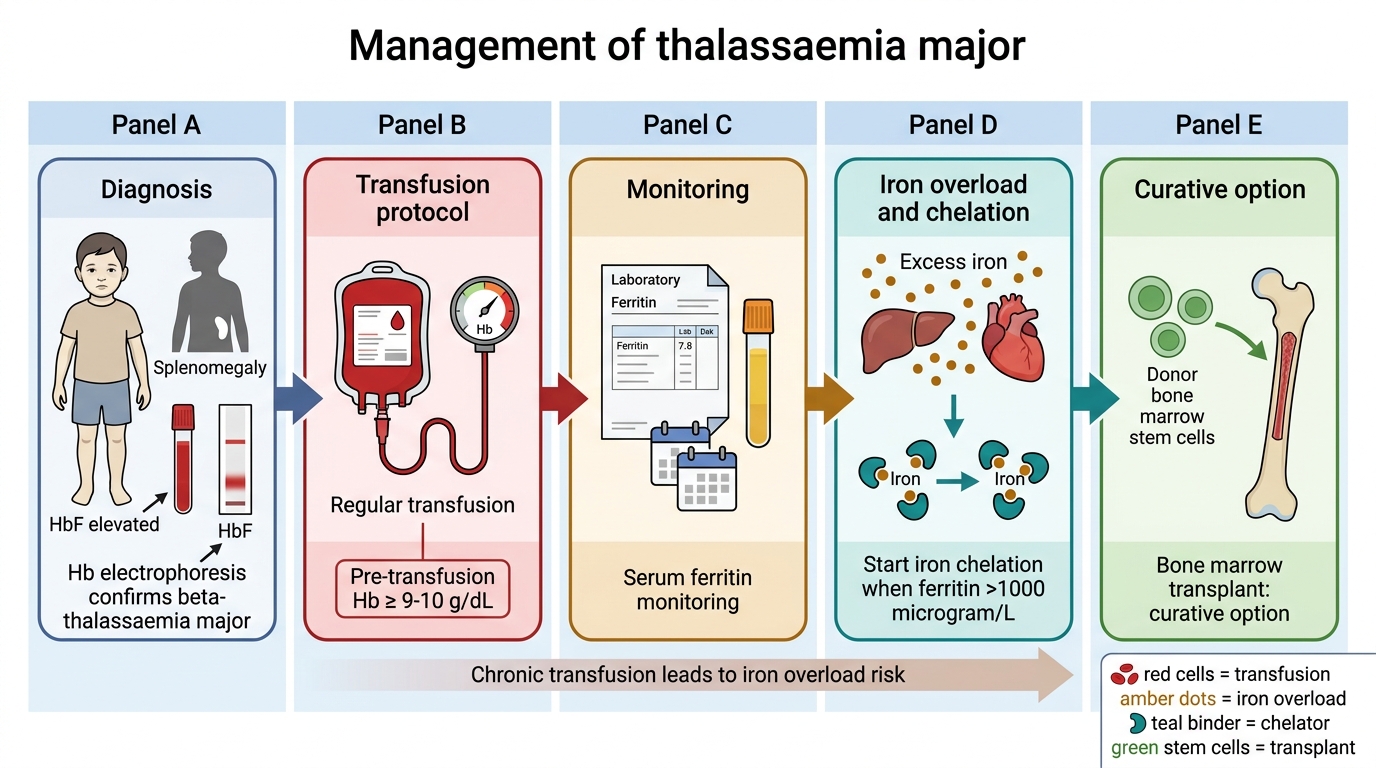
Thalassaemia Major Management Algorithm
SELF-CHECK
A 3-year-old girl with confirmed beta-thalassaemia major has been on monthly packed RBC transfusions for 18 months. Her serum ferritin is now 1350 µg/L. What is the appropriate next step?
A. Increase transfusion frequency to lower ferritin
B. Start iron chelation therapy with deferasirox or desferrioxamine
C. Perform splenectomy to reduce transfusion requirement
D. Start vitamin C supplementation to mobilise iron
Reveal Answer
Answer: B. Start iron chelation therapy with deferasirox or desferrioxamine
Serum ferritin >1000 µg/L in a transfusion-dependent child with thalassaemia major is the threshold at which iron chelation therapy must be started (typically after 10–15 transfusions or ferritin >1000 µg/L). Oral deferasirox (once daily) is currently the preferred chelating agent for compliance in children. Desferrioxamine SC infusion is effective but burdensome. Increasing transfusion frequency does not remove iron — it adds more. Splenectomy may reduce transfusion requirement in hypersplenism but is deferred until age 5–6 minimum. Vitamin C enhances iron excretion during DFO chelation but is never used alone.
Self-Assessment
Apply the haemolytic anaemia diagnostic and management framework to the following two cases before reading the analyses. The goal is to move from clinical and CBC data to a condition-specific diagnosis and then to the precise management plan for that condition — including the critical management details (chelation threshold, splenectomy age, enzyme assay timing) that distinguish a good from an excellent answer. For each case, identify the specific haemolytic disorder, state the single confirmatory diagnostic test, and outline the immediate and long-term management.
Case 1: Ramesh, a 4-year-old boy from Chhattisgarh, presents with acute pallor and swelling of both hands after a febrile illness. CBC: Hb 6.5 g/dL, MCV 78 fL, reticulocytes 5%, WBC normal, platelets normal. Peripheral smear: few sickle cells, target cells, Howell-Jolly bodies. Direct Coombs: negative.
Analysis 1:
1. Diagnosis: Sickle cell disease with dactylitis — classic presentation (infant/toddler from SCD-endemic Chhattisgarh, fever-triggered vasoocclusive crisis causing dactylitis = painful swelling of hands). Smear confirms: sickle cells, target cells, Howell-Jolly bodies (asplenia).
2. Acute management: IV hydration (prevents sickling by diluting HbS), analgesia (stepwise — paracetamol → NSAIDs → weak opioids if severe), oxygen if SpO₂ low, treat the febrile illness.
3. Long-term management: start hydroxyurea 15–20 mg/kg/day (increases HbF, reduces crisis frequency), oral penicillin V prophylaxis twice daily (functional asplenia), vaccinations (pneumococcal, meningococcal, Hib, influenza), folic acid 1 mg/day.
4. Confirm with haemoglobin electrophoresis (HbS predominant, absent HbA) and sickling test.
Case 2: Sita, a 7-year-old girl, presents with 3 days of progressive pallor and jaundice without fever, no drug exposure, no known illness in the family. Examination: mild jaundice, splenomegaly 3 cm below costal margin. CBC: Hb 7.8 g/dL, MCV 82 fL, reticulocytes 11%, platelets normal. Smear: spherocytes, no target cells, no sickle cells. Direct Coombs: positive (IgG).
Analysis 2:
1. Diagnosis: Warm AIHA — spherocytes on smear (can appear in both AIHA and HS) but DAT is POSITIVE, confirming immune mechanism. In HS, DAT is negative.
2. Management: Prednisolone 1–2 mg/kg/day orally; expect Hb response within 1–3 weeks; taper slowly after response is confirmed.
3. Important distinction: even though smear shows spherocytes, splenectomy is NOT first-line here — AIHA requires immunosuppression first; splenectomy in AIHA is reserved for refractory cases and ideally deferred in young children.
4. Investigate for underlying cause (secondary AIHA): check ANA/anti-dsDNA (SLE), EBV/CMV serology, chest X-ray (lymphoma).
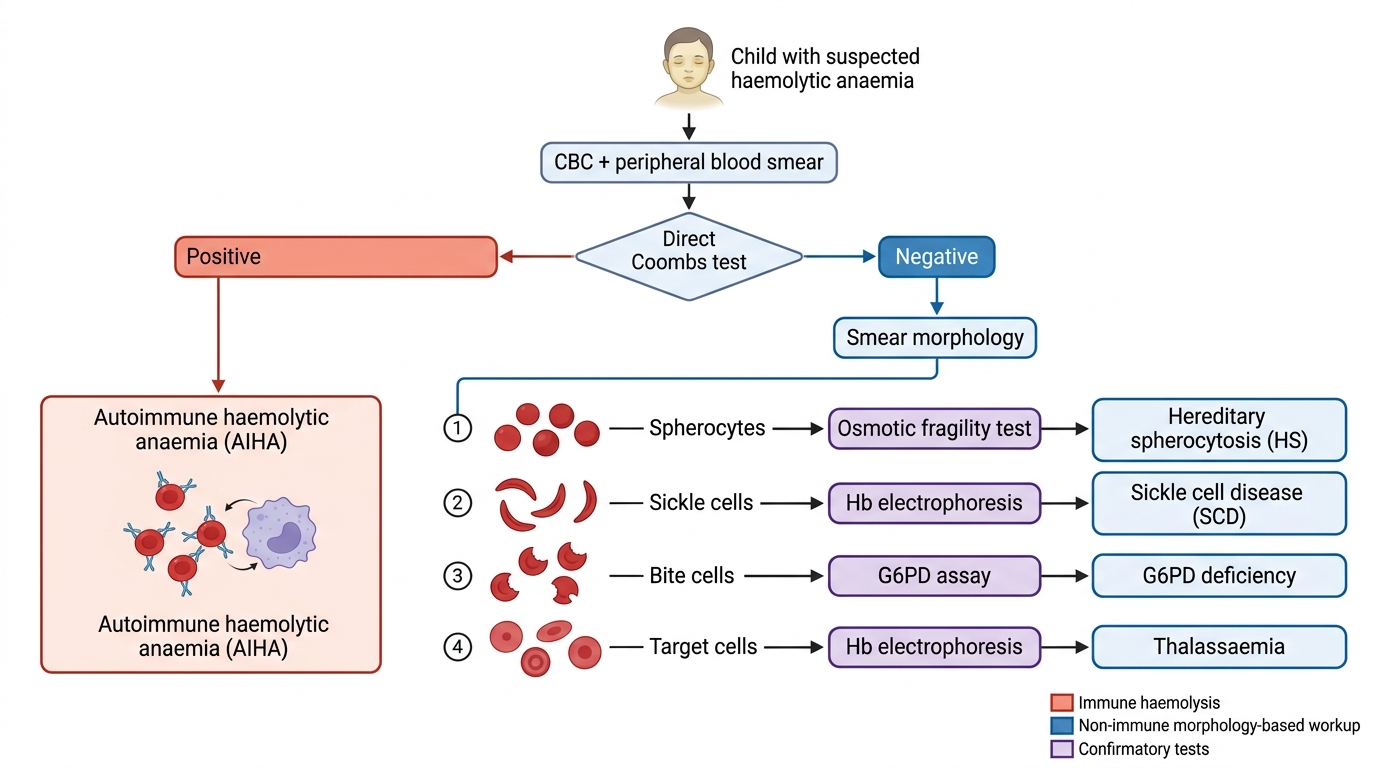
Diagnostic Algorithm for Haemolytic Anaemia in a Child