Page 13 of 34
PE26.6 | Idiopathic Thrombocytopenic Purpura — SDL Guide
Learning Objectives
- Describe the clinical features of immune thrombocytopenia (ITP) in children, including the characteristic presentation after a viral illness
- Explain the immune-mediated pathophysiology — anti-GPIIb/IIIa antibody production, splenic Fc-receptor-mediated platelet destruction, and impaired megakaryopoiesis
- Outline the diagnostic approach, including the critical distinction between ITP and acute leukaemia when hepatosplenomegaly is present
- Stratify management by severity: observation for mild disease, IVIG/anti-D/corticosteroids for moderate, and rescue therapy for life-threatening bleeding
INSTRUCTIONS
Immune thrombocytopenic purpura is the commonest cause of acute thrombocytopenia in a previously healthy child, and a paediatrician must be able to recognise it rapidly, distinguish it from sinister causes such as leukaemia, and decide whether treatment or observation is the appropriate course. The principles you learn here — platelet physiology, immune-mediated destruction, and severity-stratified care — recur across many haematological conditions you will encounter throughout your career.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 16 (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 504 (textbook)
- IAP guidelines on management of ITP in children, 2019 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 5-year-old boy is brought to the emergency department by his parents who noticed multiple small red spots appearing all over his legs and chest overnight. He had a mild cough and fever two weeks ago that resolved without treatment. He is now playful and active, with no fever. On examination you find dozens of non-blanching petechiae over the lower limbs and trunk, with a couple of small bruises on the shins. The spleen and liver are not palpable. His complete blood count returns: Hb 12.4 g/dL, WBC 7,800/µL (normal differential), platelets 6,000/µL. What is causing this dramatic drop in platelets, and is this child in danger?
WHY THIS MATTERS
Immune thrombocytopenic purpura (ITP) is the commonest cause of acute thrombocytopenia in children, responsible for the majority of paediatric presentations with petechiae and bruising in a well-looking child. Recognising ITP swiftly prevents both under-treatment (missing a life-threatening bleed) and over-investigation (performing unnecessary bone marrow biopsies on straightforward cases). Crucially, differentiating ITP from the thrombocytopenia of acute leukaemia — which can present similarly — requires a structured clinical and haematological approach. As a final-year student and future doctor, understanding this condition is directly applicable in paediatric OPD, emergency medicine, and general practice.
RECALL
Before proceeding, recall these foundations from your preclinical training:
- Platelet production: Megakaryocytes in the bone marrow shed cytoplasmic fragments (platelets); normal platelet count is 150,000–400,000/µL; lifespan ~7–10 days; major site of senescent platelet removal is the spleen via Fc-receptor-bearing macrophages.
- Haemostasis: Platelets mediate primary haemostasis — adhesion (via von Willebrand factor), activation, and aggregation to form the platelet plug at sites of endothelial injury. Coagulation factors complete secondary haemostasis.
- Bleeding phenotypes: Platelet-type (thrombocytopenia or platelet dysfunction) → petechiae, purpura, mucosal bleeding; coagulation-type (haemophilia, etc.) → ecchymoses, haemarthroses, deep muscle bleeds.
- Immune mechanisms: IgG antibodies can opsonise circulating cells for phagocytosis by splenic macrophages (Fc-receptor-mediated); this mechanism underlies several autoimmune cytopenias.
Clinical Presentation of ITP
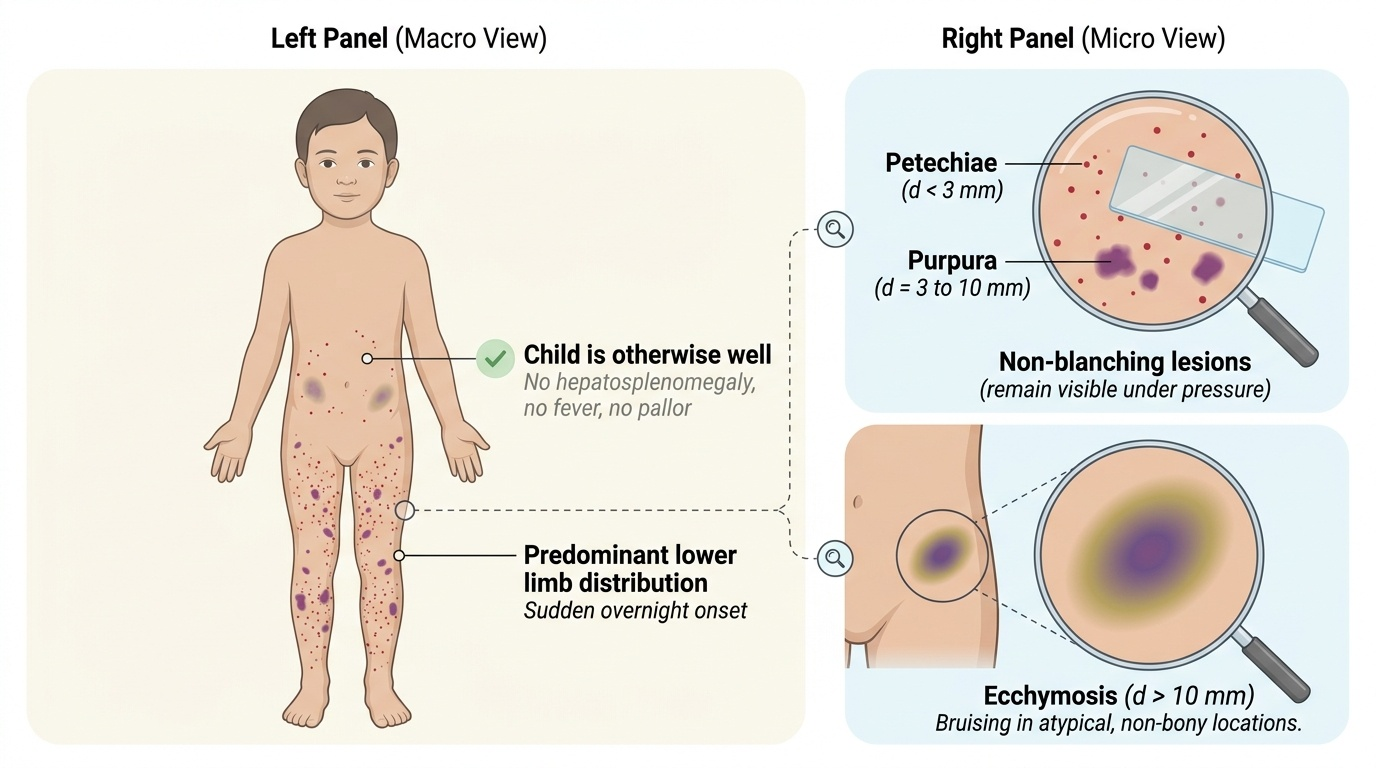
Immune thrombocytopenic purpura in children presents with a characteristic clinical pattern that, when recognised, allows confident diagnosis at the bedside. The typical patient is a child between 2 and 8 years old who presents with the sudden appearance of petechiae, purpura, and bruising over the body — often noticed by parents overnight on the child's legs. Critically, the child is otherwise well: no fever, no pallor, no significant lymphadenopathy, and — most importantly — no hepatosplenomegaly. The presence of hepatosplenomegaly should immediately shift suspicion towards an infiltrative process such as acute lymphoblastic leukaemia, and this must be excluded before attributing thrombocytopenia to ITP alone. A history of a preceding viral illness (upper respiratory tract infection, varicella, viral gastroenteritis) 2–4 weeks before the onset of bruising is present in the majority of childhood ITP cases, and this temporal association is a key clue.
Provided image
The spectrum of bleeding in ITP ranges from trivial to severe. Most children have only skin manifestations — petechiae (pinpoint, <3 mm, non-blanching haemorrhages in the skin) and purpura (larger, 3–10 mm, also non-blanching). Ecchymoses (bruises, >1 cm) may also occur, often in unusual locations like the trunk, not just bony prominences. Mucosal bleeding such as gum bleeding and epistaxis is common with platelet counts below 20,000/µL. The most feared complication is intracranial haemorrhage (ICH), which occurs in <0.5% of cases and is more likely when platelets are <10,000/µL or with head trauma. Gastrointestinal bleeding can also occur with very low counts.
The clinical classification by duration is important for management planning:
• Newly diagnosed ITP: within 3 months of diagnosis
• Persistent ITP: 3–12 months duration
• Chronic ITP: >12 months duration (commoner in adults; uncommon in children, where >80% resolve within 6–12 months)
Pathophysiology and Aetiology
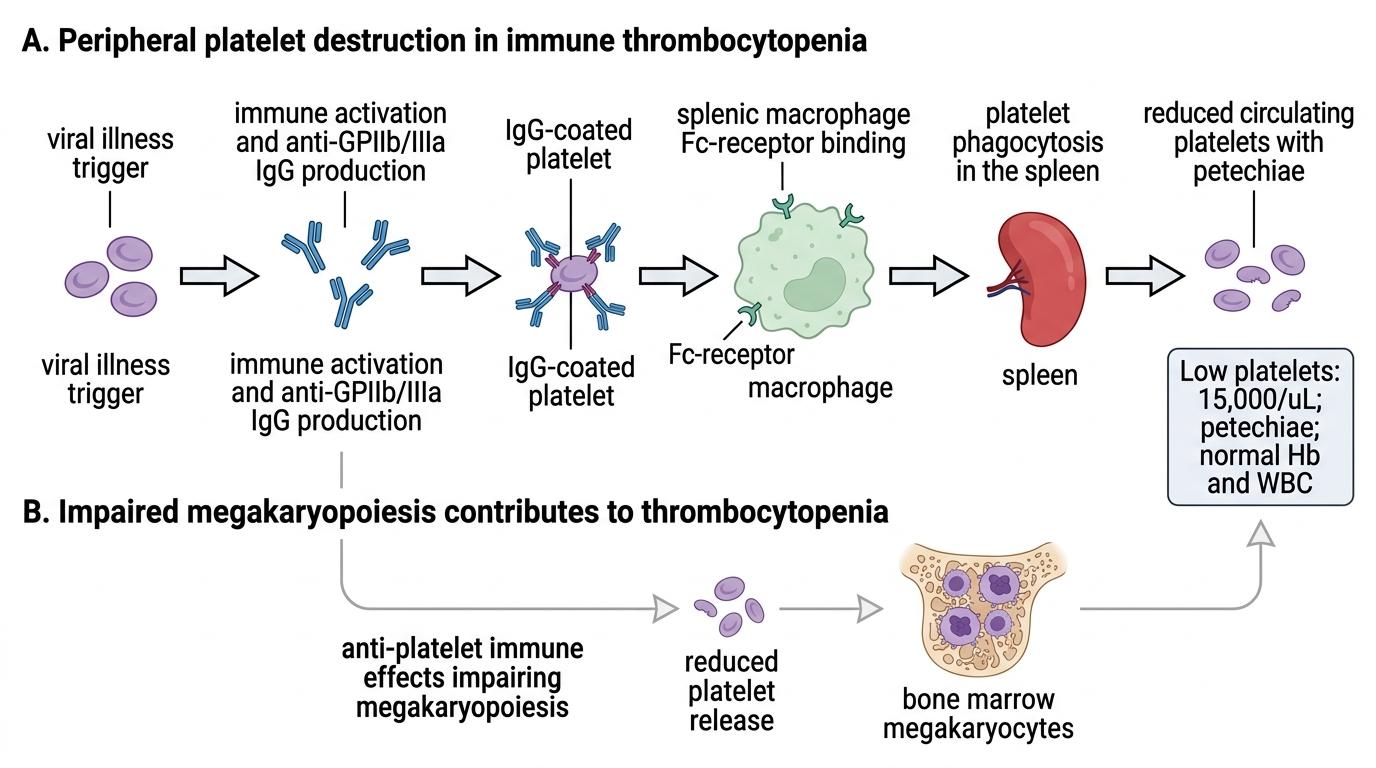
Understanding why platelets fall so dramatically in ITP requires appreciating the interplay between autoimmune antibody production, splenic macrophage activity, and impaired platelet production — three processes that all converge to produce severe thrombocytopenia. The central mechanism is the production of autoantibodies — primarily IgG antibodies directed against platelet surface glycoproteins, most commonly glycoprotein IIb/IIIa (GPIIb/IIIa, also known as integrin αIIbβ3). This complex normally mediates platelet aggregation by binding fibrinogen; when it becomes the target of autoantibodies, the antibody-coated platelets are recognised as foreign by splenic macrophages.
The sequence of events is as follows. A preceding viral infection (e.g., Epstein-Barr virus, cytomegalovirus, varicella, or non-specific URTI) triggers an immune response. By a mechanism thought to involve molecular mimicry — in which viral antigens share structural similarity with platelet glycoproteins — B-lymphocytes begin producing anti-platelet IgG. These antibodies coat circulating platelets. Splenic macrophages express Fc-γ receptors that recognise the Fc region of the bound IgG; the antibody-coated platelets are thereby phagocytosed and destroyed in the spleen. The liver (Kupffer cells) contributes additionally. Simultaneously, the autoantibodies can bind to megakaryocytes in the bone marrow, impairing platelet production — so platelet destruction is compounded by reduced production.
Vaccination (especially MMR) has also been implicated in triggering ITP through a similar mechanism, though the absolute risk is far lower than that of vaccine-preventable diseases themselves. Secondary ITP — where the thrombocytopenia is caused by an underlying disease — can occur in systemic lupus erythematosus (SLE), HIV infection, antiphospholipid syndrome, and lymphoproliferative disorders; these must be excluded in atypical presentations.
Immune Thrombocytopenia: Platelet Destruction and Impaired Production
SELF-CHECK
A 6-year-old child presents with petechiae, platelet count of 15,000/µL, normal Hb and WBC, and no hepatosplenomegaly following a viral illness 3 weeks ago. Which antibody is most commonly responsible for the platelet destruction in this condition?
A. IgM anti-platelet antibody against GPIb
B. IgG anti-platelet antibody against GPIIb/IIIa
C. IgE anti-platelet antibody against P-selectin
D. IgA anti-platelet antibody against fibrinogen
Reveal Answer
Answer: B. IgG anti-platelet antibody against GPIIb/IIIa
In ITP, IgG antibodies directed against glycoprotein IIb/IIIa (GPIIb/IIIa, integrin αIIbβ3) are the predominant pathogenic autoantibody. These antibody-coated platelets are recognised and phagocytosed by Fc-receptor-bearing splenic macrophages. IgM antibodies against GPIb can also occur but are less common. IgE and IgA anti-platelet antibodies are not the principal pathogenic mechanism in ITP.
Diagnosis and Investigation
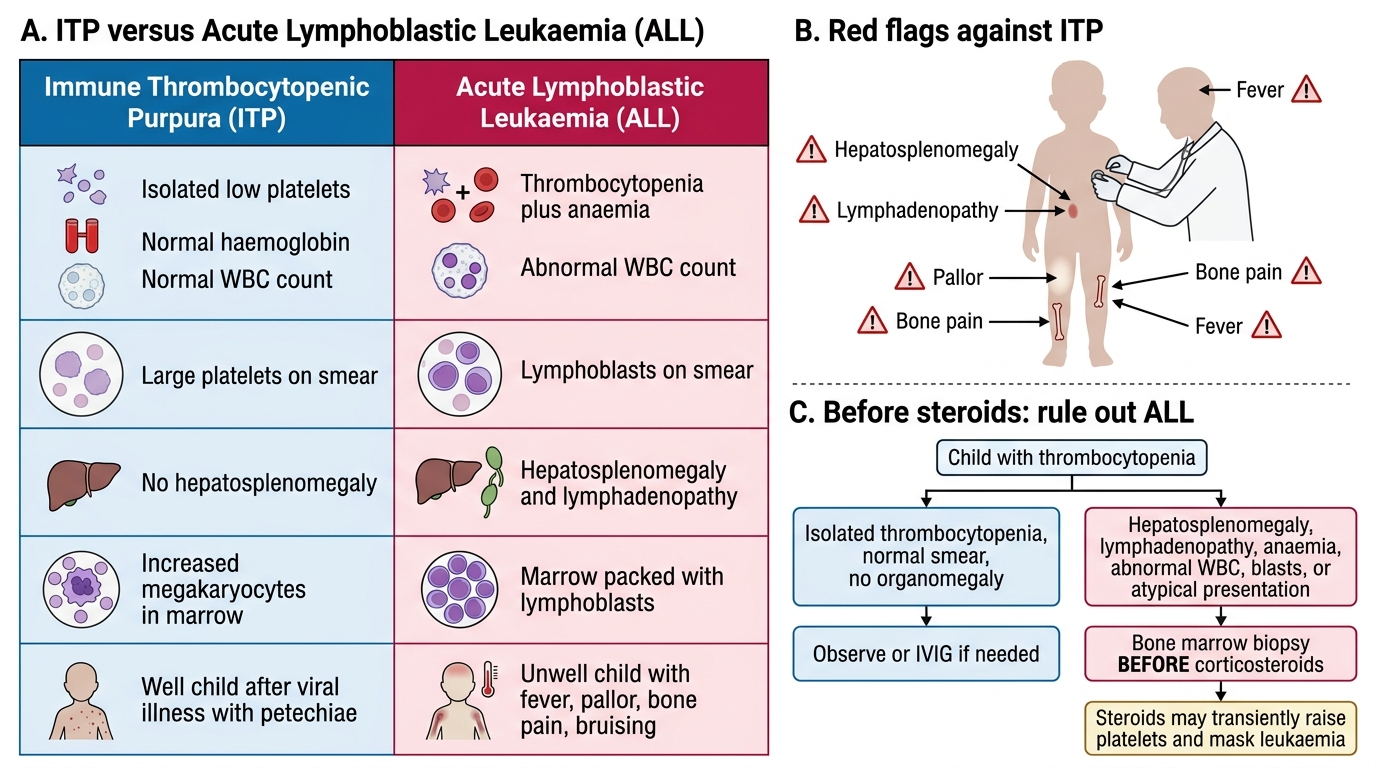
The diagnosis of ITP is a diagnosis of exclusion — there is no specific confirmatory test, and the clinician must systematically rule out other causes of thrombocytopenia before concluding that a child has ITP. The first and most critical investigation is the complete blood count (CBC). In ITP, the defining haematological pattern is isolated thrombocytopenia: a low platelet count with a normal haemoglobin, normal white cell count, and normal differential. Any abnormality in the non-platelet lineages — anaemia, leucopenia, or leucocytosis — must prompt a bone marrow examination to exclude leukaemia or aplastic anaemia. This single point is the most important differentiator between ITP and acute leukaemia at the bedside.
The peripheral blood smear is mandatory and provides essential information. In ITP, platelets are reduced in number but those present are often larger than normal (megathrombocytes), reflecting increased thrombopoietic drive in the marrow. Importantly, red cell morphology is normal (no fragments/schistocytes, which would suggest thrombotic thrombocytopenic purpura or haemolytic-uraemic syndrome), and blast cells are absent. The presence of blast cells on the smear is an absolute indication for bone marrow biopsy.
Bone marrow examination is NOT routinely required for typical childhood ITP (post-viral, isolated thrombocytopenia, well child). It is indicated in atypical presentations: when steroids are planned before the diagnosis is confirmed (steroids alone could mask leukaemia), when the CBC shows cytopenias beyond thrombocytopenia alone, when hepatosplenomegaly is present, when the child looks systemically unwell, or when the platelet count fails to respond to standard therapy. Additional tests to consider for secondary causes include ANA (for SLE), HIV serology in at-risk patients, and antiphospholipid antibody screen (for recurrent cases). Coagulation tests (PT, aPTT) are normal in ITP — platelet-type bleeding does not disturb coagulation pathway tests.
| Investigation | Finding in ITP | Significance |
|---|---|---|
| CBC | Platelets ↓↓, Hb normal, WBC normal | Isolated thrombocytopenia |
| Peripheral smear | Reduced platelets, megathrombocytes; no blasts, no red cell fragments | Excludes TTP/HUS/leukaemia |
| Bone marrow (if indicated) | Megakaryocytes ↑ or normal, no blast infiltration | Excludes aplastic anaemia, leukaemia |
| PT/aPTT | Normal | Confirms platelet-type (not coagulation-type) bleeding |
| ANA, anti-dsDNA | Positive in SLE-related thrombocytopenia | Excludes secondary ITP |
| HIV serology | Positive in HIV-associated thrombocytopenia | Excludes secondary ITP |
ITP versus ALL: Key Diagnostic Differences
CLINICAL PEARL
The hepatosplenomegaly trap: The single most dangerous error in suspected ITP is initiating corticosteroid therapy in a child who actually has acute lymphoblastic leukaemia. Steroids will transiently improve the platelet count in both conditions — masking the leukaemia diagnosis for days to weeks and delaying potentially curative treatment. The rule is: if hepatosplenomegaly, lymphadenopathy, anaemia, or abnormal white cells are present on CBC or smear, or if the presentation is atypical, perform a bone marrow biopsy BEFORE starting steroids. A normal-looking child with isolated thrombocytopenia after a viral illness can be observed or treated with IVIG, which does not obscure leukaemia.