Page 38 of 48
PE27.12 | Duchenne Muscular Dystrophy — SDL Guide (Part 2)
Management: Slowing Progression and Supportive Care
Management of DMD is multifaceted, targeting disease progression, respiratory and cardiac function, orthopaedic complications, nutrition, psychological wellbeing, and quality of life across the entire lifespan. The last two decades have seen DMD begin to transition from a purely supportive condition — where clinicians could only manage complications as they arose — to one with disease-modifying therapies that meaningfully alter the natural history. Corticosteroids now extend ambulation by 2–5 years; mutation-specific gene therapies are changing outcomes for defined patient subgroups; cardiac and respiratory prophylaxis strategies have substantially improved survival. This transformation means that management must now begin proactively in the ambulatory phase, long before complications emerge, following the structured care framework described in the 2018 Lancet Neurology DMD Care Considerations. A critically important rule that applies to every pharmacological intervention in this module: all drug doses in children must be calculated on a weight-based mg/kg basis — never extrapolate from or use adult fixed doses for a growing child.
1. Corticosteroids — the standard of care for slowing progression:
Corticosteroids are the only proven standard therapy that delays loss of ambulation and preserves respiratory and cardiac function in DMD. The mechanism is thought to involve anti-inflammatory, membrane-stabilising, and muscle-regenerative effects. Recommended agents and doses:
• Prednisolone (prednisone): 0.75 mg/kg/day orally (weight-based; maximum ~40 mg/day) — continuous daily regimen is most evidence-based.
• Deflazacort: 0.9 mg/kg/day orally — similar efficacy, possibly less weight gain than prednisolone, but causes more cataracts.
Corticosteroids extend ambulation by 2–5 years and preserve respiratory function. They are typically started at age 4–5 years (once the child is in the plateau phase of motor development — before the decline begins), continued throughout the ambulatory phase and into the non-ambulatory phase for pulmonary and cardiac benefits.
Side effects requiring monitoring: weight gain (caloric restriction counselling), bone loss (calcium 500–1000 mg/day + vitamin D 400–1000 IU/day + DEXA monitoring), growth suppression, behavioural changes, cataracts (deflazacort > prednisone), hypertension, glucose intolerance.
2. Gene-targeted mutation-specific therapies (precision medicine):
• Exon-skipping antisense oligonucleotides (ASOs): Convert out-of-frame to in-frame deletions, restoring partial (Becker-like) dystrophin production. Examples: eteplirsen (exon 51 skipping — targets ~13% of DMD patients with amenable mutations), golodirsen (exon 53), viltolarsen (exon 53), casimersen (exon 45). Given by weekly IV infusion. These are approved in the US/EU for specific exon amenability.
• Stop-codon readthrough: Ataluren (translarna) — small molecule that promotes ribosomal readthrough of premature stop codons; indicated for nonsense mutation DMD (~13% of patients); approved in Europe.
• Mini-dystrophin gene therapy (AAV-based): Phase 3 trials underway (delandistrogene moxeparvovec — Elevidys — FDA approved June 2023 for 4–5 years with confirmed DMD mutation, ambulatory); delivers a truncated but functional mini-dystrophin gene.
3. Cardiac management:
• Echocardiogram annually from age 10 (or earlier if symptoms).
• ACE inhibitors (enalapril 0.1 mg/kg/day, titrated) or ARBs — start prophylactically at age ~10 years regardless of cardiac symptoms (evidence shows they slow cardiomyopathy progression).
• Beta-blockers and aldosterone antagonists added as cardiomyopathy progresses.
• Anticoagulation if severely dilated heart with poor EF.
4. Respiratory management:
• Annual spirometry; FVC <50% predicted → consider BiPAP/NIV.
• Respiratory physiotherapy — assisted cough techniques (cough-assist device) for secretion clearance.
• Influenza and pneumococcal vaccination (higher risk of respiratory infections).
5. Orthopaedic management:
• Ankle-foot orthoses (AFO) for equinus deformity; physiotherapy for contractures.
• Surgical Achilles tendon release in carefully selected ambulatory patients.
• Scoliosis surveillance (radiograph) annually; surgical fusion when Cobb angle >30–40° in non-ambulatory patients.
6. Family counselling and genetic services:
• Carrier testing for maternal relatives — female carrier detection by genetic testing (not by CK alone, which is insensitive).
• Prenatal diagnosis (chorionic villus sampling, amniocentesis) for future pregnancies.
• Psychological support for family; connect to DMD patient advocacy groups (Parent Project Muscular Dystrophy).
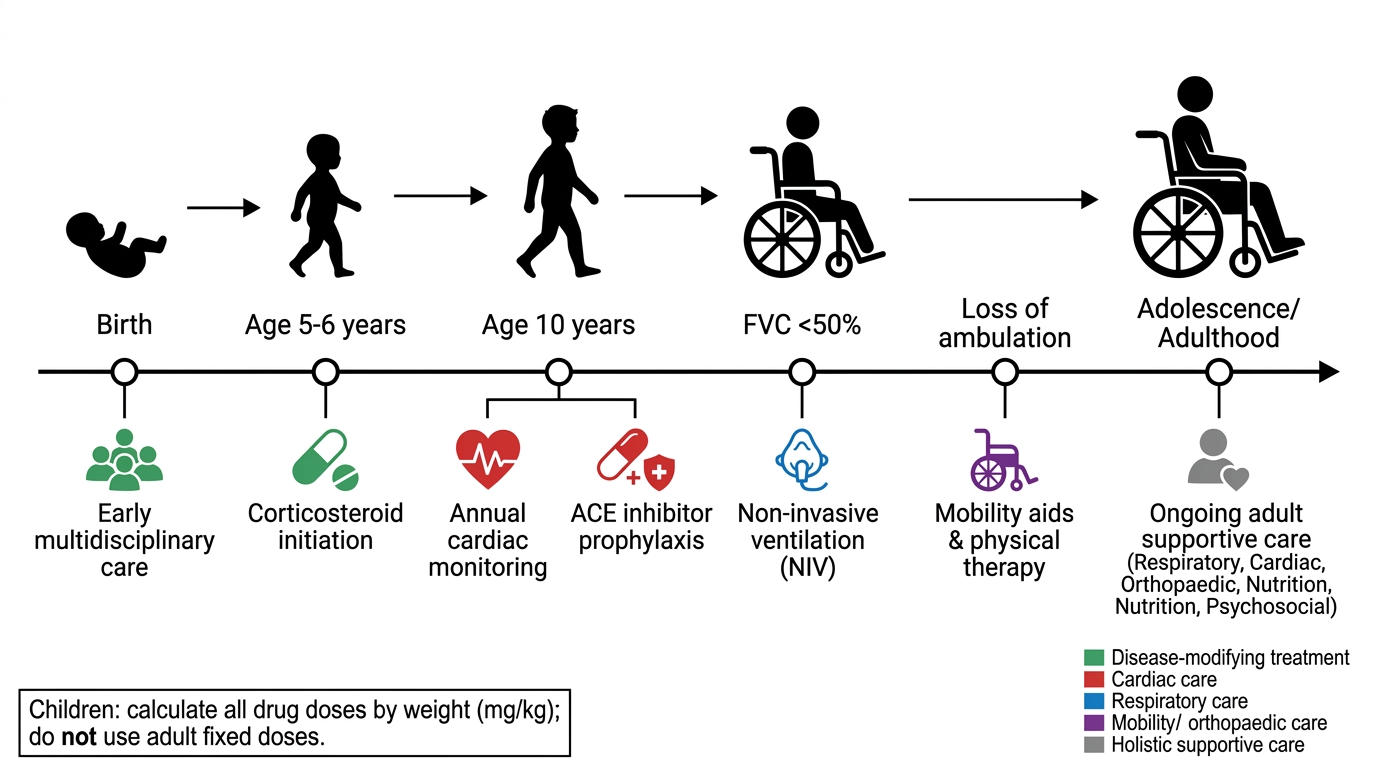
DMD Management Timeline: Birth to Adulthood
CLINICAL PEARL
Two critical points that are frequently examined and clinically vital: First, calf pseudohypertrophy is NOT true muscle hypertrophy — the enlarged calves in DMD are made of fat and fibrous tissue replacing necrotic muscle fibres. They look big but test WEAK. This is the paradox that confuses families ('his legs look so strong!') and distinguishes DMD from a well-muscled child. Second, corticosteroid dosing in children is ALWAYS weight-based — prednisolone 0.75 mg/kg/day, deflazacort 0.9 mg/kg/day. Never state or prescribe an adult fixed dose for a child. Additionally, CK is highest early in the disease (when muscle mass is abundant but being rapidly destroyed) and actually falls in later non-ambulatory stages as muscle is replaced by fibrous tissue — a declining CK in late-stage DMD does NOT mean improvement; it means there is less muscle left to destroy.
SELF-CHECK
A 6-year-old boy with confirmed DMD (exon 48-50 deletion, out-of-frame) is started on prednisolone. His weight is 20 kg. Which statement about his steroid therapy is CORRECT?
A. The standard dose is 40 mg/day (adult fixed dose) — the same for all children with DMD
B. The standard dose is 0.75 mg/kg/day = 15 mg/day for this child; corticosteroids delay loss of ambulation by 2-5 years
C. Deflazacort should not be used because it causes more weight gain than prednisolone
D. Corticosteroids are not indicated in DMD until the child loses ambulation, as they have no proven benefit in the ambulatory phase
Reveal Answer
Answer: B. The standard dose is 0.75 mg/kg/day = 15 mg/day for this child; corticosteroids delay loss of ambulation by 2-5 years
Corticosteroid dosing in children is ALWAYS weight-based. Prednisolone standard dose = 0.75 mg/kg/day; for a 20 kg child = 15 mg/day. This is the evidence-based dose that delays ambulation loss by 2–5 years and preserves respiratory/cardiac function — started in the ambulatory phase (typically age 4–6) BEFORE decline begins, not after. Deflazacort (0.9 mg/kg/day) is an alternative with similar efficacy and possibly less weight gain (though more cataracts). Adult fixed doses are NEVER appropriate for paediatric dosing.
Self-Assessment: Duchenne Muscular Dystrophy
Use these structured case vignettes and concept questions to consolidate your understanding of DMD across the full clinical arc — from recognising the presentation to planning the diagnostic workup and explaining therapeutic options to families. Examination questions on DMD frequently target the reading-frame rule, the CK interpretation, and the management of comorbidities. Work through each scenario independently before reviewing the key concepts below, as active recall at this stage is more effective than passive re-reading.
Case vignette 1: A 4-year-old boy is brought with frequent falls and difficulty climbing stairs for the past 6 months. His mother noticed that he walks on his toes and his calves look 'very muscular.' On examination, he has prominent, firm calves, lumbar lordosis, waddling gait, and Gower's sign. DTRs are present but diminished. CK is 22,000 U/L.
1. What is the most likely diagnosis? What is the inheritance pattern?
2. What single genetic investigation would you request to confirm the diagnosis?
3. What age would you start corticosteroids, and what dose for a 16 kg child?
4. What cardiac monitoring would you initiate?
Case vignette 2: A family is referred for genetic counselling. The proband is a 7-year-old boy with DMD (confirmed exon 50 deletion, out-of-frame). The boy's maternal aunt is 30 years old and pregnant.
1. What is the aunt's risk of being a carrier?
2. What investigation would confirm carrier status?
3. What prenatal diagnostic options are available?
Key concepts checklist:
• DMD = X-linked recessive; dystrophin gene Xp21; out-of-frame mutation → absent dystrophin.
• Clinical hallmarks: age 3–5 years; Gower's sign; calf pseudohypertrophy (fatty/fibrous, NOT true muscle); toe-walking; proximal > distal weakness.
• CK markedly elevated: 10–100x ULN (5,000–50,000 U/L in early disease); DECREASES late when muscle is replaced.
• Diagnosis: MLPA (first-line for deletions/duplications) → NGS for point mutations; muscle biopsy if inconclusive.
• DMD vs Becker: out-of-frame mutation → absent dystrophin → severe DMD; in-frame → truncated protein → milder Becker.
• Corticosteroids: prednisolone 0.75 mg/kg/day OR deflazacort 0.9 mg/kg/day — weight-based, started before decline (~age 4–6).
• Cardiac: ACE inhibitor prophylaxis from ~age 10; annual echo.
• Respiratory: FVC <50% → BiPAP; cough-assist device.
• Gene therapies: exon skipping (eteplirsen, golodirsen), ataluren (stop-codon), mini-dystrophin gene therapy (Elevidys).