Page 26 of 33
PE13.4 | Calcium Magnesium Nutrition — SDL Guide (Part 2)
Calcium Homeostasis: PTH, Vitamin D, and Phosphate Interplay
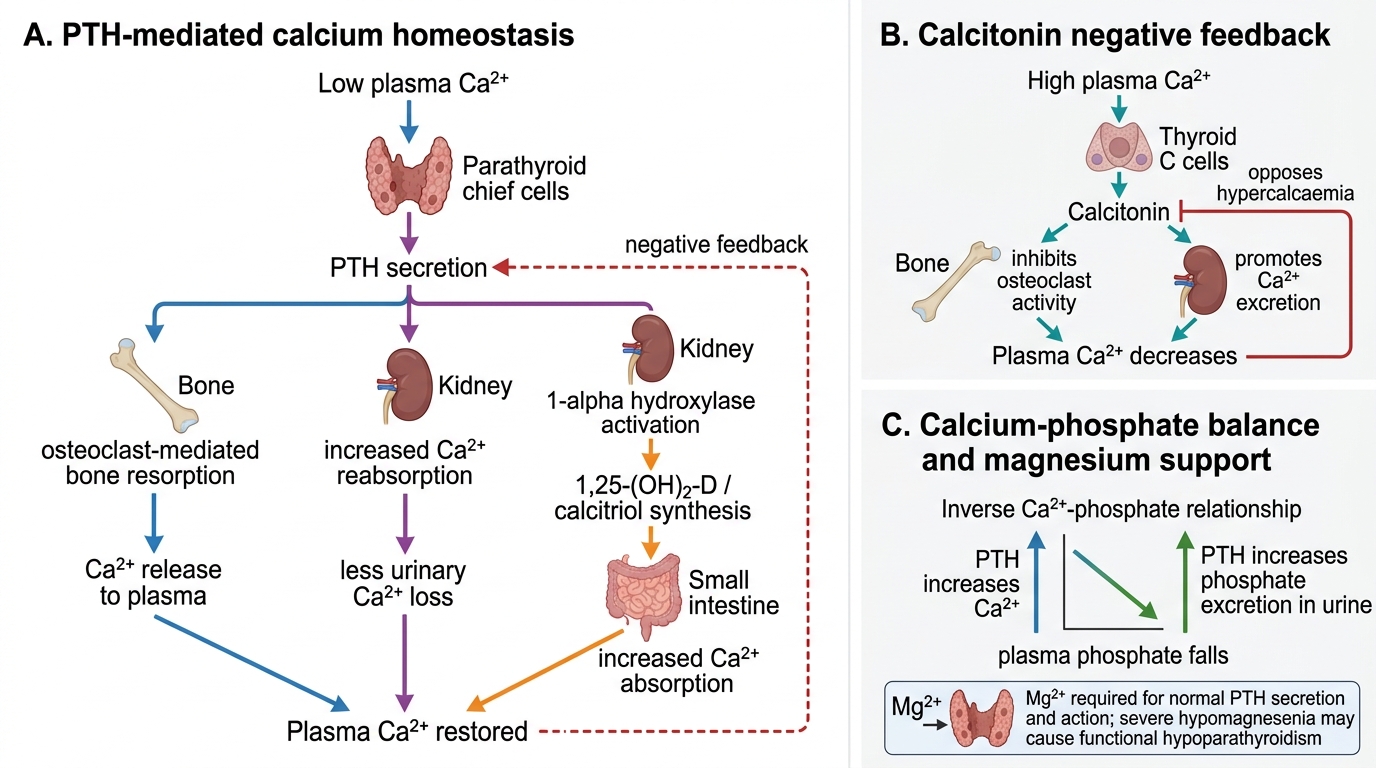
Calcium homeostasis is maintained within a narrow serum range (8.5–10.5 mg/dL) by a tightly regulated hormonal axis involving parathyroid hormone (PTH), 1,25-dihydroxyvitamin D (calcitriol), and calcitonin. The central sensing mechanism is the calcium-sensing receptor (CaSR) on parathyroid chief cells, which detects minute falls in ionised serum calcium and triggers PTH secretion within seconds. PTH is the dominant acute-phase regulator: it acts on three target organs simultaneously — bone, kidney, and the intestine (indirectly via vitamin D) — to restore serum calcium over a timescale ranging from minutes (bone) to hours (kidney) to days (intestinal). This multi-organ, multi-timescale architecture means that defects anywhere in the axis — deficiency of vitamin D, failure of renal 1α-hydroxylase, resistance at the target receptor, or impaired PTH secretion from magnesium depletion — all converge on the same final pathway: falling ionised calcium, rising neuromuscular excitability, and the risk of tetany or seizure.
The three-pronged action of PTH in response to hypocalcaemia:
1. Bone: PTH binds osteoblasts, which secondarily activate osteoclasts, releasing calcium and phosphate into the extracellular fluid. This is the fastest buffer — response in minutes to hours.
2. Kidney: PTH increases calcium reabsorption in the distal convoluted tubule, simultaneously causes phosphaturia (preventing a rise in serum phosphate from bone dissolution), and activates renal 1α-hydroxylase — converting 25-OH-D (calcidiol) to 1,25-OH2-D (calcitriol).
3. Intestine (indirect, via calcitriol): 1,25-OH2-D upregulates calbindin-D9k and TRPV6 channels in enterocytes, increasing both transcellular (active) calcium absorption and phosphate absorption. This is the slowest limb — taking days.
Calcitonin (from thyroid parafollicular C-cells) is secreted in response to hypercalcaemia. It inhibits osteoclast activity and decreases renal tubular calcium reabsorption. Calcitonin is physiologically important in neonates and pregnant/lactating women but is of lesser importance in adult calcium homeostasis.
The inverse calcium–phosphate relationship is a crucial clinical concept in neonatal hypocalcaemia. In the extracellular fluid, calcium and phosphate concentrations are constrained by a solubility product. An abrupt rise in serum phosphate — as occurs when a newborn is fed unmodified cow's milk, which contains three times the phosphate of breast milk — drives ionised calcium down, precipitating late-onset neonatal hypocalcaemia. This is the mechanism behind "cow's milk hypocalcaemia" in the first week of life.
Vitamin D deficiency disconnects the intestinal limb of the axis: without adequate calcitriol, intestinal calcium absorption falls, serum calcium tends to drift downward, and there is a compensatory rise in PTH (secondary hyperparathyroidism). In early vitamin D deficiency, PTH successfully maintains serum calcium at the expense of bone mineral — producing radiological rachitic changes before hypocalcaemia becomes overt. In severe or prolonged deficiency, however, serum calcium eventually falls, triggering frank hypocalcaemic tetany superimposed on rickets.
Calcium Homeostasis: PTH, Calcitriol, Calcitonin and Phosphate
Magnesium Homeostasis and Its Role in Calcium Regulation
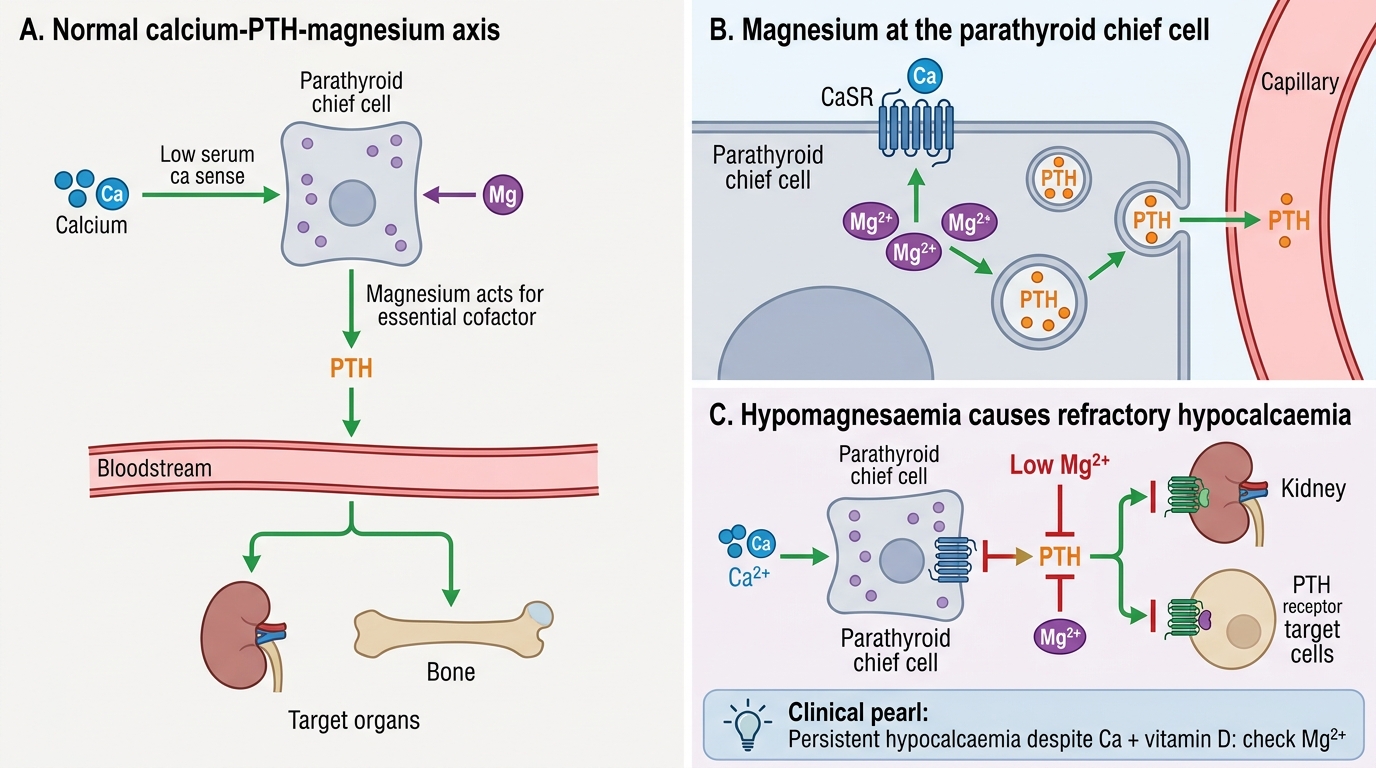
Magnesium functions as an obligate cofactor for more than 300 enzymatic reactions, the majority involving adenosine triphosphate (ATP). The biologically active form of ATP in all kinase and phosphotransferase reactions is Mg-ATP2−, the complex of magnesium chelated to the ATP molecule's γ-phosphate. This means that magnesium is essential for glycolysis, the citric acid cycle, fatty acid oxidation, DNA replication, RNA transcription, and ribosomal protein synthesis — effectively for all energy-requiring cellular processes. Magnesium is also required for the activation of adenylyl cyclase and the function of sodium–potassium ATPase, explaining many of its neuromuscular effects when depleted.
The critical interface between magnesium and calcium homeostasis operates at two levels:
Level 1 — PTH secretion: Parathyroid chief cells require intracellular magnesium for the exocytosis of PTH-containing secretory granules. In hypomagnesaemia, the signalling pathway linking the calcium-sensing receptor to PTH vesicle release is impaired, resulting in a paradoxically low or inappropriately normal PTH level despite hypocalcaemia. This is termed functional hypoparathyroidism induced by magnesium depletion.
Level 2 — End-organ PTH responsiveness: Even if some PTH is secreted, its downstream actions depend on G-protein-coupled receptor signalling, which requires Mg2+ as a cofactor for the G-protein (Gs) α-subunit GTPase cycle. Severe hypomagnesaemia therefore creates both a secretory and a receptor-level resistance to PTH.
The clinical consequence is refractory hypocalcaemia — a serum calcium that cannot be corrected by calcium or vitamin D supplementation alone, in a child with co-existing hypomagnesaemia. The management principle is clear: replace magnesium first; the calcium will follow. Oral magnesium replacement (magnesium oxide or glycinate) is used for mild deficiency. For symptomatic or severe hypomagnesaemia, IV magnesium sulphate is given at 25–50 mg/kg IV over 30–60 minutes (maximum 2 g per dose), with cardiac monitoring.
Homeostatic control of serum magnesium involves: dietary absorption from the small intestine (predominantly the ileum), bone as the reservoir, and renal excretion regulated in the thick ascending limb of the loop of Henle (TRPM6/7 channels) and the distal convoluted tubule. Kidneys are the primary regulator of magnesium balance and can reduce fractional excretion to near-zero during deficiency — but they cannot compensate for ongoing losses from the gut.
Magnesium, PTH Function, and Refractory Hypocalcaemia
CLINICAL PEARL
The refractory hypocalcaemia trap: A child with persistent hypocalcaemia that does not correct after 48–72 hours of adequate calcium gluconate infusion and vitamin D supplementation should prompt an urgent serum magnesium measurement. Hypomagnesaemia is frequently overlooked because it does not produce dramatic symptoms of its own in early stages — the presentation is dominated by the hypocalcaemia. The mechanism is dual: impaired PTH secretion from parathyroid chief cells AND reduced end-organ sensitivity to PTH at the receptor level (both require Mg2+ for G-protein signalling). Replacing magnesium (IV MgSO4 25–50 mg/kg over 30–60 min, max 2 g/dose) is the key intervention — the calcium will correct within hours once Mg is repleted. Common clinical settings in paediatrics: prolonged diarrhoea or acute gastroenteritis, short-bowel syndrome, primary intestinal hypomagnesaemia (genetic TRPM6 mutations), and chronic diuretic therapy.
Calcium Deficiency: Clinical Features, Rickets, and Tetany
Calcium deficiency in paediatric practice manifests across a spectrum from the seizing neonate to the toddler with bowing legs to the adolescent with pathological fractures. The two main clinical syndromes are hypocalcaemic tetany (and its extreme, hypocalcaemic seizures) and nutritional rickets. Understanding the distinction between these presentations — and between their subcategories — is essential for correct diagnosis and management. Neonatal hypocalcaemia, for instance, is not a single entity: early-onset (within 72 hours) and late-onset (after day 4–7) forms have different aetiologies, different biochemical profiles, and different responses to treatment. Similarly, nutritional rickets must be distinguished from the hereditary form (X-linked hypophosphataemic rickets) whose management is entirely different. Hypocalcaemic tetany can present subtly as jitteriness in a neonate or dramatically as laryngospasm in an infant — and in both cases, the clinician must also consider whether co-existing hypomagnesaemia is driving refractory hypocalcaemia before concluding that calcium supplementation alone is sufficient.
Provided image
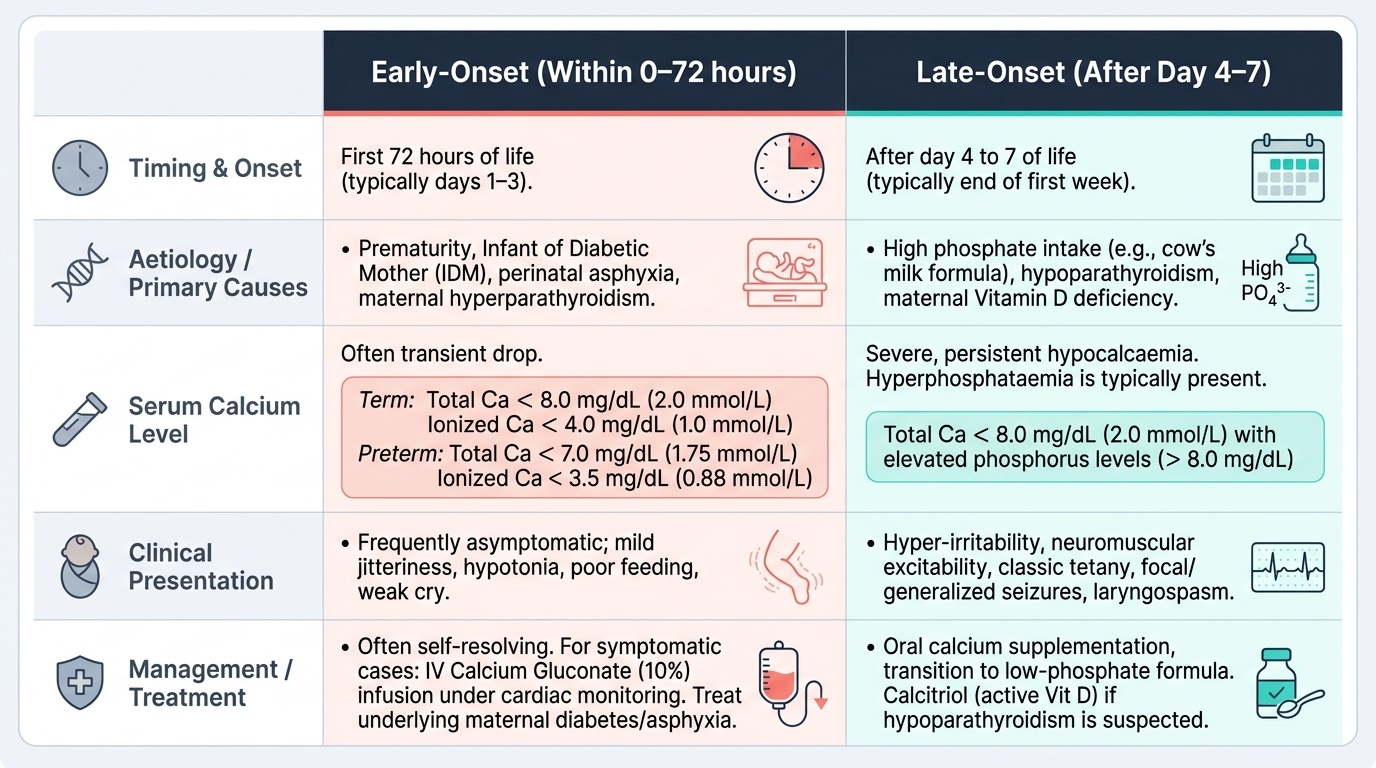
Neonatal Hypocalcaemia: Defined as serum total calcium <8 mg/dL (or ionised calcium <4 mg/dL [<1.0 mmol/L]) in term neonates, or total calcium <7 mg/dL (ionised <3.5 mg/dL [<0.88 mmol/L]) in preterm neonates. There are two distinct forms:
| Feature | Early-onset (within 0–72 h) | Late-onset (after day 4–7) |
|---|---|---|
| Aetiology | Prematurity, IDM (maternal hyperglycaemia → foetal hyperinsulinism → hypomagnesaemia), perinatal asphyxia | High-phosphate feeds (unmodified cow's milk), hypomagnesaemia, maternal vitamin D deficiency |
| PTH response | Immature parathyroid → inadequate PTH surge at birth | Phosphate load overwhelms PTH capacity; may be compounded by Mg deficiency |
| Clinical features | Jitteriness, high-pitched cry, apnoea, seizures | Irritability, tetany, seizures |
| Management | IV calcium gluconate (1–2 mL/kg of 10% solution, slow IV), cardiac monitoring; then oral calcium | IV calcium gluconate for acute seizures; switch to breast milk or low-phosphate formula; address Mg if needed |
Hypocalcaemic Tetany: Tetany is the most dramatic manifestation of hypocalcaemia in older infants and children, caused by lowered threshold for neuromuscular excitability as ionised calcium falls below ~1.1 mmol/L. Clinical signs include:
• Trousseau sign: carpal spasm (flexion of wrist and MCP joints with extension of IP joints and adduction of thumb — "obstetrician's hand") induced by inflating a blood-pressure cuff above systolic for 3 minutes. This is the most sensitive clinical test for latent tetany.
• Chvostek sign: ipsilateral facial muscle twitch on tapping the facial nerve just anterior to the ear. Less specific — positive in ~10–25% of normocalcaemic individuals.
• Laryngospasm: potentially life-threatening — inspiratory stridor, "crowing" respirations; occurs preferentially in infants.
• Generalised tonic-clonic seizures: may be the presenting event, particularly in neonates and infants.
Emergency management of hypocalcaemic tetany or seizure: IV calcium gluconate 10% 1–2 mL/kg (maximum 20 mL) given slowly over 5–10 minutes with continuous cardiac monitoring (risk of bradycardia and cardiac arrest with rapid infusion). Follow with a maintenance calcium infusion (30–75 mg/kg/day of elemental calcium) until the patient can be switched to oral supplementation.
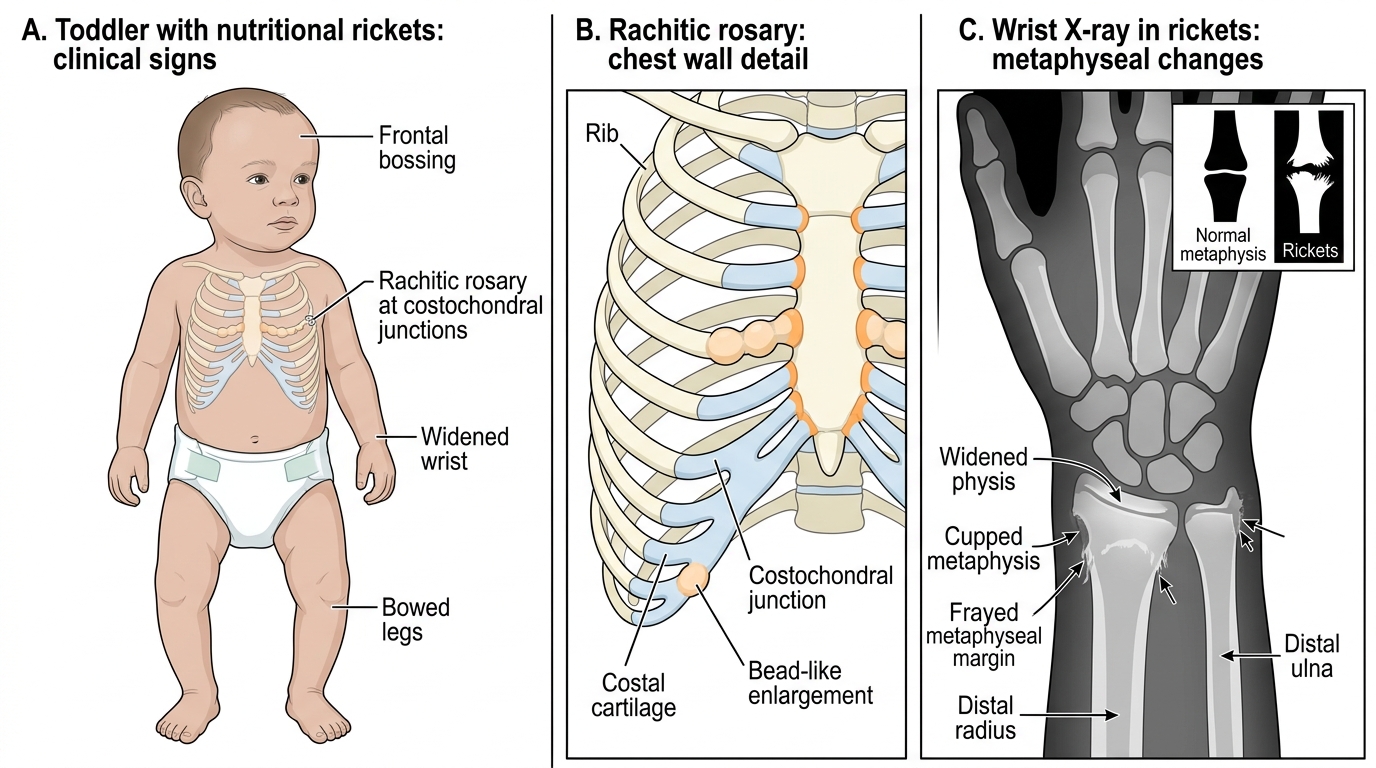
Nutritional Rickets: In India, nutritional rickets is caused most commonly by combined vitamin D and calcium deficiency; pure calcium-deficiency rickets (without vitamin D deficiency) also occurs in toddlers fed exclusively cereal-based diets with low dairy intake and adequate sun exposure. The characteristic features include:
• Craniotabes: pinging sensation on pressing the occiput/parietal bones (softening of the cranial vault — seen in infants <6 months).
• Frontal bossing and widened anterior fontanelle beyond 18 months.
• Rachitic rosary: palpable beading at costochondral junctions from expanded unmineralised osteoid.
• Harrison's sulcus: horizontal groove along the lower chest where the diaphragm attaches, from compliant, softened ribs.
• Widened wrists and ankles (broadened metaphyses on palpation).
• Bowing of legs (genu varum) in toddlers who have started weight-bearing, or genu valgum (knock-knees) in older children.
• X-ray changes: cupping, fraying, and splaying of metaphyses; widened growth plate; reduced bone density.
• Biochemistry: low/normal serum calcium, low phosphate, elevated alkaline phosphatase, elevated PTH, low 25-OH-D (<20 ng/mL).
Distinguish nutritional rickets from X-linked hypophosphataemic (XLH) rickets: XLH is caused by PHEX gene mutations → elevated FGF23 → renal phosphate wasting; bowing legs despite normal calcium and vitamin D; serum phosphate low, serum Ca normal, 25-OH-D normal; treatment requires phosphate supplementation + calcitriol (NOT vitamin D alone). Never treat XLH with high-dose vitamin D.
Management of nutritional rickets: IAP/2022 recommendations — Vitamin D 2000–4000 IU/day for 3 months (or stoss/shock dose 1000 IU/kg/day for 10 days, max 400,000 IU single dose for severe deficiency), combined with oral calcium supplementation 500–1000 mg/day of elemental calcium. Follow-up X-rays and alkaline phosphatase at 3 months to confirm healing.
Clinical and Radiological Features of Nutritional Rickets
SELF-CHECK
A 3-week-old term neonate, exclusively fed unmodified cow's milk, presents with a brief generalised seizure. Serum calcium is 6.8 mg/dL, serum phosphate is 8.5 mg/dL, PTH is low-normal. Which of the following best explains the pathophysiology?
A. Vitamin D deficiency reducing intestinal calcium absorption
B. High phosphate load from cow's milk precipitating calcium, reducing ionised serum calcium
C. Immature parathyroid glands failing to respond to hypocalcaemia
D. Primary hypoparathyroidism (DiGeorge syndrome)
Reveal Answer
Answer: B. High phosphate load from cow's milk precipitating calcium, reducing ionised serum calcium
This is a classic presentation of late-onset neonatal hypocalcaemia. Unmodified cow's milk contains approximately 3 times the phosphate content of breast milk. When a neonate is fed cow's milk, the high phosphate load is absorbed from the gut and raises serum phosphate. Due to the inverse Ca-phosphate solubility relationship in extracellular fluid, this suppresses ionised calcium. The PTH response may be inadequate to compensate. Vitamin D deficiency (option A) can cause hypocalcaemia but takes weeks to months to manifest and is less acute. Immature parathyroid glands (option C) is the mechanism for EARLY-onset neonatal hypocalcaemia within 72 hours, not late-onset. DiGeorge syndrome (option D) is a possibility but is a diagnosis of exclusion in this context.