Page 2 of 17
PA15.1 | Vitamin B12 & Folate Metabolism — Deficiency Pathogenesis — SDL Guide (Part 2)
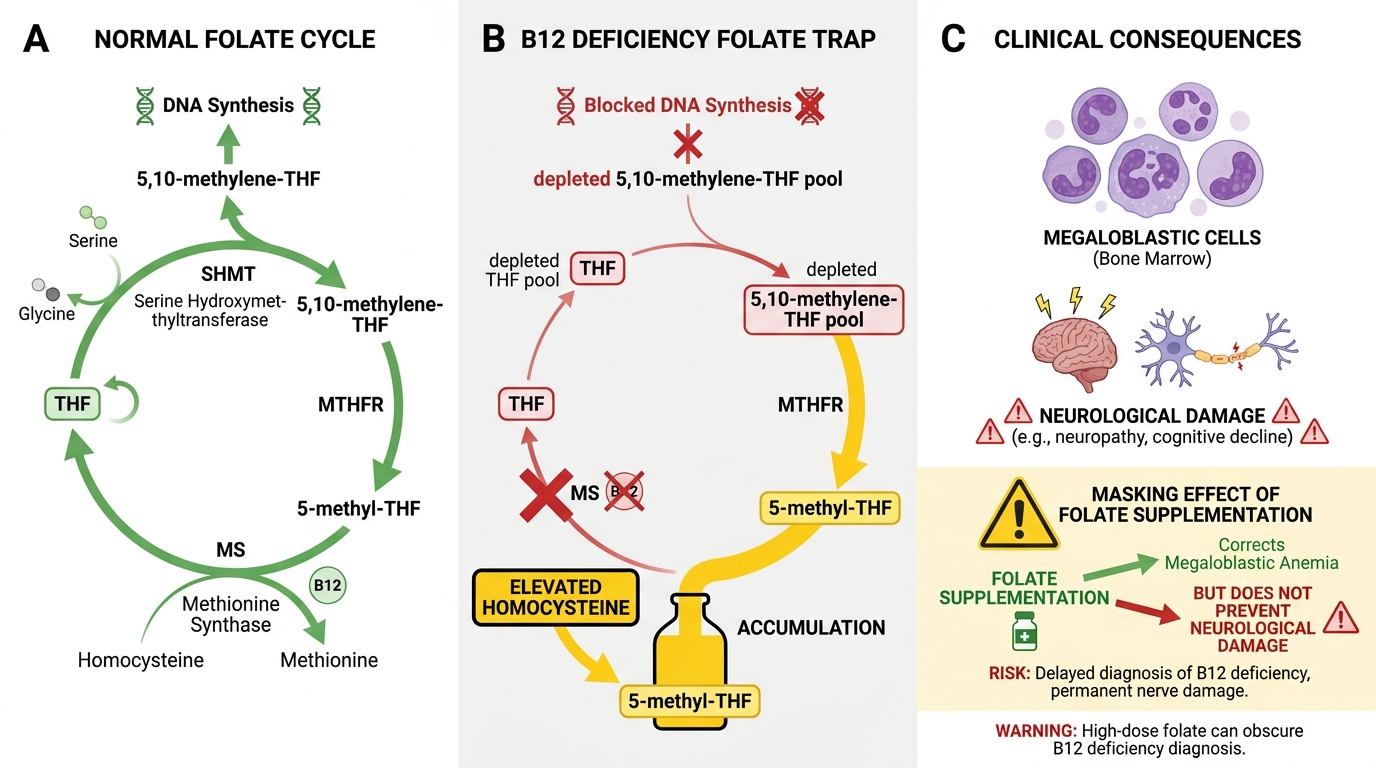
The Folate Trap — Why B12 Deficiency Starves Cells of Folate
The Folate Trap in B12 Deficiency: Mechanism and Clinical Impact
This is the pivotal concept linking B12 deficiency to megaloblastosis — even when dietary folate intake is normal.
The trap explained:
- In the methionine-synthase reaction, 5-methyl-THF donates its methyl group to homocysteine and is demethylated to THF.
- Without B12, methionine synthase cannot work. 5-methyl-THF piles up; it cannot be converted to THF.
- Cells cannot synthesise 5,10-methylene-THF (needed for thymidylate synthesis) or 10-formyl-THF (needed for purine synthesis).
- DNA synthesis is blocked — specifically, thymidine monophosphate (dTMP) cannot be made.
This state — a functional intracellular folate deficiency despite normal serum folate — is the "folate trap" (also called the methylfolate trap).
Key insight for exams: giving folate supplements to a B12-deficient patient partially bypasses the trap through an alternative (DHFR-independent) pathway, temporarily correcting the blood picture. But it does NOT restore MUT activity. The neurological damage from B12 deficiency continues silently — this is the dangerous "masking" effect of folate supplementation in undiagnosed B12 deficiency.

The Folate Trap in B12 Deficiency
SELF-CHECK
A patient with undiagnosed B12 deficiency is prescribed folic acid supplements by a general practitioner. What is the most dangerous consequence of this action?
A. Folic acid will worsen the anaemia by competing with B12 for absorption
B. Folic acid partially corrects anaemia but neurological damage from B12 deficiency continues undetected
C. Folic acid raises methylmalonic acid levels, masking the B12 assay
D. Folic acid inhibits methionine synthase, deepening the metabolic block
Reveal Answer
Answer: B. Folic acid partially corrects anaemia but neurological damage from B12 deficiency continues undetected
Folic acid partially bypasses the folate trap, restoring enough one-carbon units for DNA synthesis so the blood count improves. However, it cannot substitute for B12 in the methylmalonyl-CoA mutase reaction. Myelin-related methylation defects persist, and subacute combined degeneration progresses silently. This is why the NMC and WHO guidelines require ruling out B12 deficiency before starting folic acid supplementation.
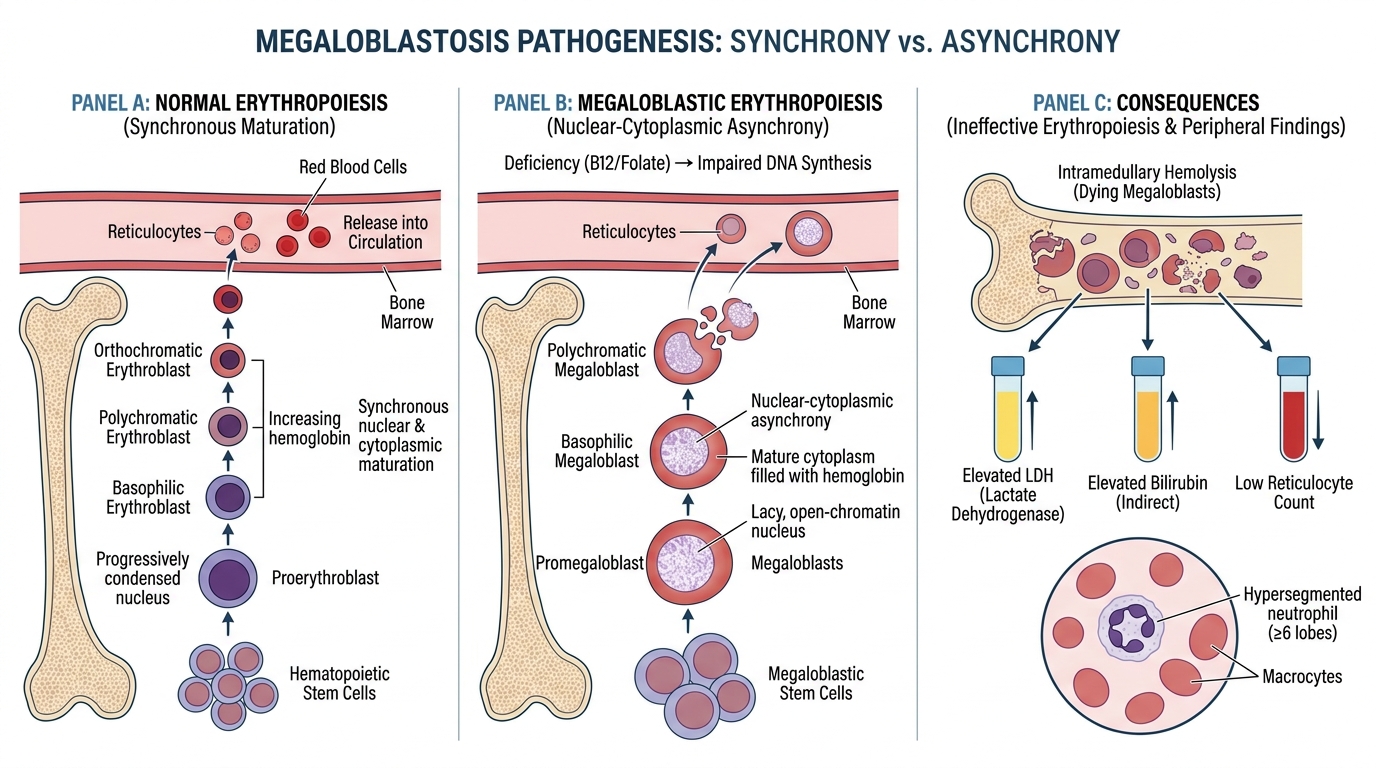
Pathogenesis of Megaloblastosis — Nuclear-Cytoplasmic Asynchrony
Pathogenesis of Megaloblastosis: Nuclear-Cytoplasmic Asynchrony
All rapidly dividing cells — bone marrow precursors, GI epithelium, oral mucosa — are vulnerable to DNA synthesis blockade.
Nuclear-cytoplasmic asynchrony is the defining pathological event:
- The nucleus cannot divide (blocked dTMP → blocked DNA replication) so the cell cannot complete mitosis.
- The cytoplasm, however, continues to mature normally: haemoglobin accumulates, the cell grows larger.
- The result is a large cell with an immature, lacy, open-chromatin nucleus that looks disproportionately young for the amount of cytoplasm present — the hallmark of a megaloblast.
Ineffective erythropoiesis: most megaloblasts die within the marrow before reaching peripheral blood (intramedullary haemolysis). This is why:
• Serum LDH is markedly elevated (cell death releases LDH).
• Serum indirect bilirubin is mildly elevated (haemoglobin breakdown).
• The reticulocyte count is low despite abundant marrow precursors — the precursors die before maturing.
The same asynchrony affects granulocyte precursors → hypersegmented neutrophils (≥5 lobes in >5% of neutrophils) appear in peripheral blood — often the earliest finding on a blood film, sometimes before macrocytosis is evident.

Megaloblastic Erythropoiesis: Bone Marrow Aspirate Findings
CLINICAL PEARL
The haematologist's shortcut: A hypersegmented neutrophil (≥5 lobes) on a blood smear is the most sensitive early sign of megaloblastic anaemia — it appears before the MCV rises and before symptoms are clinically obvious. Train your eye to look for it reflexively whenever you see macrocytosis. Conversely, a truly macrocytic anaemia WITHOUT any hypersegmented neutrophils should make you reconsider the diagnosis (liver disease? hypothyroidism? drugs?).
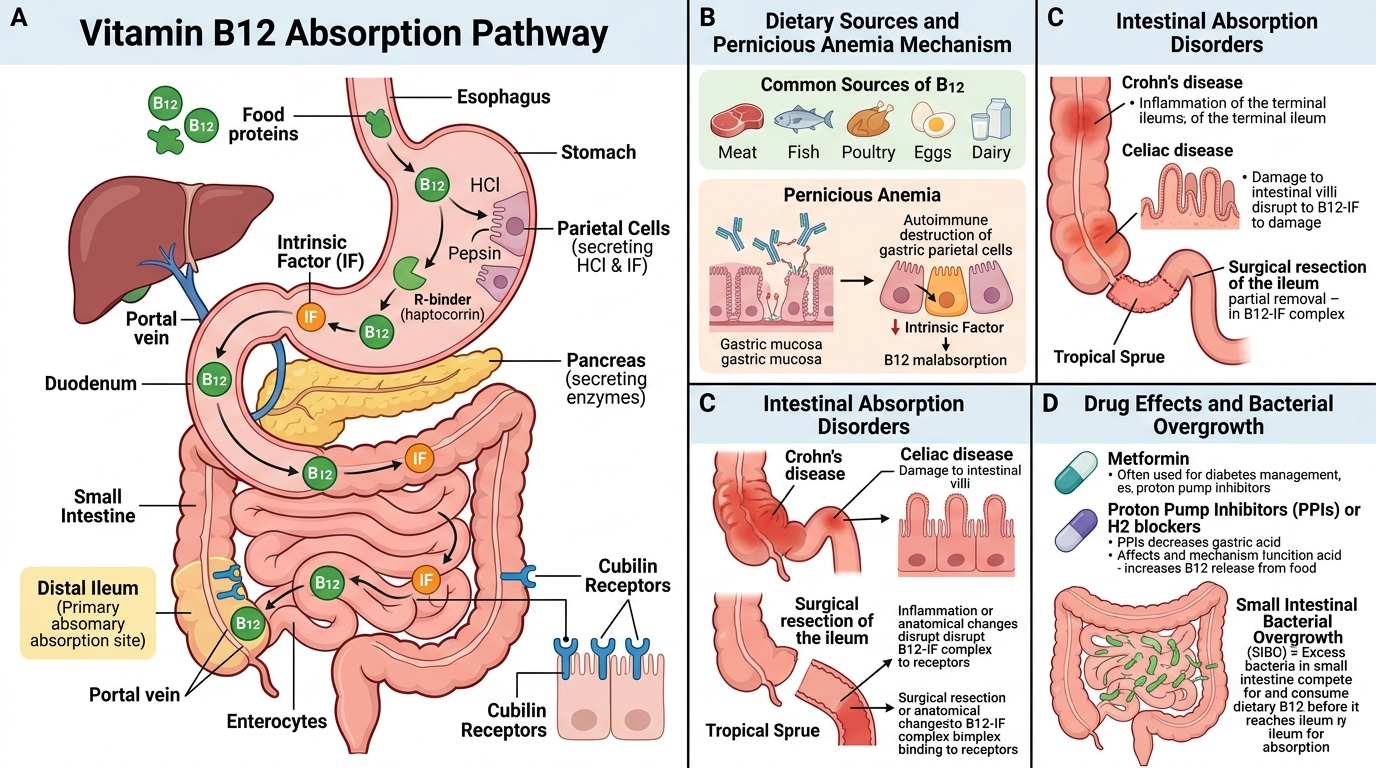
Causes of Vitamin B12 Deficiency
Vitamin B12 Absorption Pathway and Causes of Deficiency
Organise by step in the absorption pathway — it makes the mechanism self-evident:
1. Inadequate dietary intake
• Strict vegans (lacto-ovo vegetarians are partially protected).
• Severe malnutrition (rare as an isolated cause in adults; commoner in exclusively breastfed infants of vegan mothers).
2. Impaired gastric secretion of intrinsic factor
• Pernicious anaemia (PA) — autoimmune destruction of gastric parietal cells; most common cause in adults >50 years. Two antibody types: anti-parietal-cell antibodies (90% sensitive, less specific) and anti-intrinsic-factor antibodies (60% sensitive, highly specific). Associated with other autoimmune diseases (Hashimoto's, Addison's, vitiligo).
• Total/partial gastrectomy (surgical removal of IF-secreting tissue).
• Chronic atrophic gastritis (autoimmune or H. pylori–induced).
3. Impaired intestinal absorption
• Ileal disease or resection (Crohn's disease, TB ileitis, >60 cm resection).
• Bacterial overgrowth (blind loop syndrome) — bacteria consume luminal B12 before cubilin uptake.
• Diphyllobothrium latum (fish tapeworm) — worm competes for luminal B12 (endemic in Scandinavia and parts of India near large freshwater fish lakes).
• Pancreatic insufficiency — failure to cleave haptocorrin by pancreatic proteases (rare but important mechanism).
4. Drugs and toxins
• Nitrous oxide (N₂O, anaesthetic gas/recreational use) irreversibly oxidises B12 from its active (reduced) to inactive (oxidised) form; can precipitate acute B12 deficiency neuropathy in hours in borderline-deficient patients.
• Metformin (long-term) — reduces ileal cubilin expression; subtle B12 reduction in ~10–30% of patients.
• Prolonged proton-pump inhibitor (PPI) use — impairs acid-dependent release of protein-bound B12.
5. Functional deficiency
• Transcobalamin II deficiency (rare congenital disorder) — B12 cannot be transported to tissues despite normal absorption.