Page 6 of 21
PA16.2 | Sickle Cell Disease & Thalassaemia — Hereditary Haemolytic Anaemias — SDL Guide (Part 2)
Study the smear above carefully. Note that sickle cells (drepanocytes) are elongated with tapered pointed ends — not simply 'curved'. Target cells have a central dense spot, pale ring, and peripheral dense rim. Howell-Jolly bodies are small, singular, deeply stained inclusions within RBCs; their presence on a film is virtually diagnostic of a functionally or surgically asplenic patient.
Distinguishing feature on smear review: In SCD, both ISCs (permanently sickled) and reversibly sickled forms may be present. The ISC count correlates with disease severity — more ISCs = more chronic endothelial damage.

Sickle Cell Disease: Peripheral Blood Smear and HbS Polymerization
The diagram above captures the molecular cascade. Key teaching points:
1. The trigger is deoxygenation — clinical situations that promote crises (cold, infection, dehydration, high altitude) work by promoting deoxygenation or increasing MCHC.
2. Hydroxyurea increases HbF synthesis, diluting the HbS pool and reducing polymerisation — this is the molecular basis of the most effective medical therapy in SCD.
3. Each sickling–unsickling cycle progressively oxidises and cross-links the membrane skeleton, culminating in the irreversibly sickled cell even when oxygenated.

HbS Polymerization and RBC Sickling Process
Thalassaemia: Pathogenesis and Classification
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
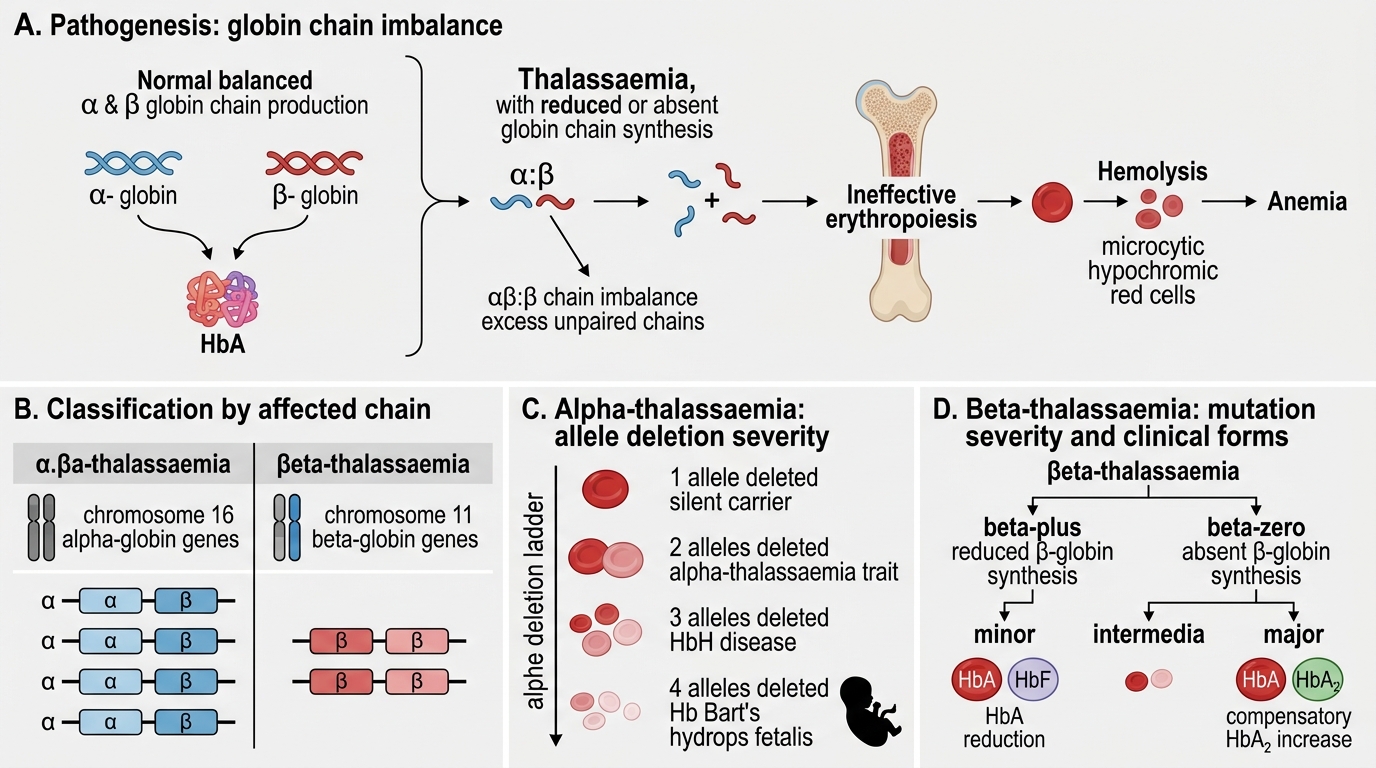
Thalassaemia: Pathogenesis and Classification
Thalassaemias are a heterogeneous group of disorders caused by quantitative reduction or absence of structurally normal globin chain synthesis. The result is a stoichiometric imbalance between α and β chains.
Classification by affected chain:
| Type | Defect | Chromosome | Genetics |
|---|---|---|---|
| α-Thalassaemia | ↓/absent α-globin | 16 | Gene deletions (4 α-gene alleles) |
| β-Thalassaemia | ↓/absent β-globin | 11 | Point mutations (2 β-gene alleles) |
α-Thalassaemia severity by allele deletion:
- 1 allele deleted (α-/αα): Silent carrier — normal indices
- 2 alleles deleted (α-/α- or --/αα): α-Thalassaemia trait — mild microcytic, hypochromic anaemia
- 3 alleles deleted (--/α-): HbH disease — HbH (β4 tetramers) form, moderately severe haemolytic anaemia
- 4 alleles deleted (--/--): Hb Bart's hydrops fetalis — incompatible with postnatal life; γ4 tetramers (Hb Bart's) have extremely high O2 affinity
β-Thalassaemia classification:
- β0: Complete absence of β-chain synthesis (most severe mutations)
- β+: Reduced (partial) β-chain synthesis
- Thalassaemia minor (β-thal trait, heterozygous β0 or β+): Mild anaemia, microcytosis — clinically resembles IDA but ferritin is normal
- Thalassaemia intermedia: Moderately severe, not transfusion-dependent at baseline
- Thalassaemia major (Cooley's anaemia, homozygous β0/β0 or β0/β+): Severe, transfusion-dependent from first year of life