Page 5 of 16
AS8.3 | Pharmacologic Management of Pain — SDL Guide
CLINICAL SCENARIO
A 52-year-old man is recovering from a right hemicolectomy for colon cancer. On post-operative day 1 he has an NRS pain score of 8/10 despite having received intravenous morphine 4 mg an hour ago. The junior resident reaches for 'more morphine' — but the anaesthesiologist on the acute pain service asks three rapid questions instead: Has he been given any paracetamol? Is there an NSAID prescribed? Is the epidural running correctly? The answers are no, no, and 'we ran out of the infusion.' Within an hour — after paracetamol 1 g IV, diclofenac 75 mg PR, and reconnection of the epidural — his pain score is 3/10 and he is beginning to mobilise. This scenario illustrates the fundamental principle of modern analgesic pharmacology: no single drug, regardless of its potency at one receptor, matches the efficacy, safety, and functional recovery provided by a rationally combined multimodal regimen. Understanding the mechanism, receptor pharmacology, clinical indications, dose ranges, and adverse-effect profiles of each analgesic class — and understanding how they complement one another — is the clinical pharmacology that every anaesthesiologist must command.
WHY THIS MATTERS
Analgesic pharmacology is one of the highest-stakes domains in clinical medicine. Undertreated acute pain drives physiological harm: tachycardia, hypertension, increased myocardial oxygen demand, impaired respiratory mechanics (post-thoracotomy splinting), reduced mobility, and immune suppression. It also increases the risk of transition to chronic pain through central sensitisation. Conversely, over-reliance on opioids without multimodal co-prescribing contributes to respiratory depression, delirium, ileus, urinary retention, nausea, and — at a population level — opioid misuse and dependence. The anaesthesiologist's role is to design and titrate the analgesic regimen that maximally controls pain with minimal adverse effects. This requires granular pharmacological knowledge: not just that 'paracetamol, NSAIDs, and opioids work' but precisely how each drug interacts with its target, what the dose-response relationship looks like, what the ceiling effects are, and what patient-specific factors modify the risk-benefit calculation. NMC 2024 competency AS8.3 requires final-year students to describe the pharmacology and use of drugs in pain management — a foundation upon which every clinical decision in acute pain, palliative care, and chronic pain practice rests.
RECALL
Recall from pharmacology and physiology: receptor pharmacology distinguishes agonists (activate receptor, produce effect), antagonists (bind receptor, block effect), and partial agonists (submaximal intrinsic activity). Opioids act on G-protein-coupled receptors; activation of Gi/o proteins reduces cAMP, decreases Ca²⁺ conductance, and increases K⁺ conductance — the net effect is reduced neuronal excitability. NSAIDs inhibit COX-1 and COX-2 enzymes, reducing prostaglandin synthesis; COX-2 is predominantly inducible at inflammatory sites, whereas COX-1 is constitutive (gastric cytoprotection, platelet thromboxane). The spinal cord dorsal horn contains μ, δ, and κ opioid receptors; the periaqueductal grey (PAG) contains μ receptors — exogenous opioids produce analgesia at both sites. These concepts are the pharmacological scaffolding for this module.
The WHO Analgesic Ladder: Structure, Rationale, and Clinical Application
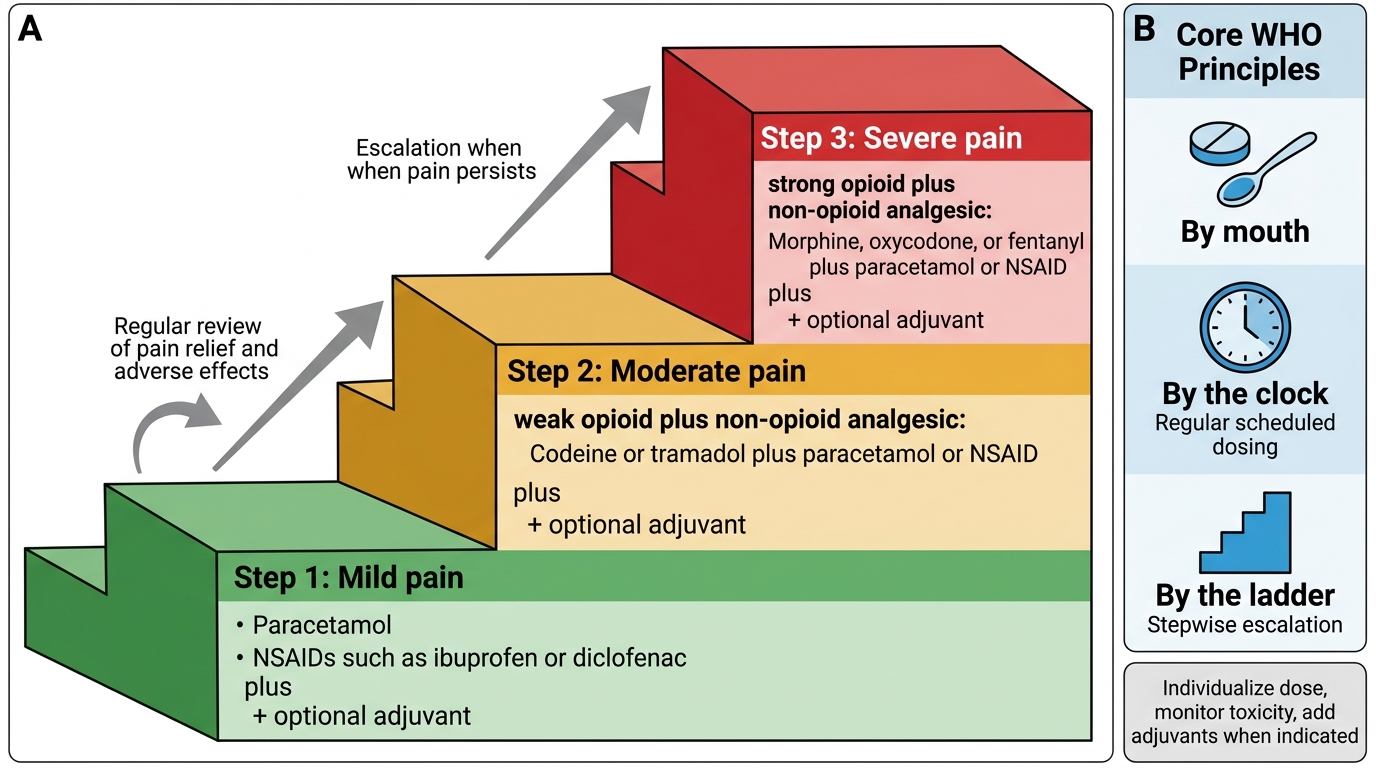
The WHO analgesic ladder, introduced in 1986 for cancer pain management and subsequently adopted for acute and chronic pain, provides the conceptual framework organising analgesic therapy. It describes a stepwise escalation approach: begin at the step appropriate to the patient's pain severity, not necessarily at the bottom.
Step 1 — Non-opioid analgesics: paracetamol (acetaminophen) and/or NSAIDs, with or without adjuvant drugs (e.g., corticosteroids, tricyclic antidepressants, gabapentinoids). Indicated for mild pain (NRS 1–3).
Step 2 — Weak opioids ± non-opioids ± adjuvants: codeine, tramadol, or low-dose oral morphine. For moderate pain (NRS 4–6). Some guidelines have collapsed steps 1 and 2, using low-dose strong opioids instead of weak opioids, given the variable efficacy and poor adverse-effect profile of codeine (a prodrug requiring CYP2D6 metabolism to active morphine — highly variable across populations).
Step 3 — Strong opioids ± non-opioids ± adjuvants: morphine, oxycodone, hydromorphone, fentanyl, buprenorphine. For severe pain (NRS 7–10). The guiding principle at this step is dose titration to effect — there is no ceiling dose for strong opioids in malignant pain, but titration must be balanced against adverse effects.
The revised WHO ladder for palliative care and some modern perioperative frameworks add a fourth conceptual step — interventional analgesia (spinal opioids, nerve blocks, implanted devices) — for pain uncontrolled by systemic pharmacotherapy.
The ladder's core principle — 'by mouth, by the clock, by the ladder' — translates into three clinical rules:
1. Prefer the oral route whenever possible (least invasive, patient-controlled, avoids injection pain and infection risk).
2. Prescribe regular dosing 'by the clock' (not 'as needed' for background pain) to maintain steady-state plasma levels and prevent pain breakthrough.
3. Prescribe a rescue dose for breakthrough pain: typically 1/6 to 1/10 of the total daily opioid dose, available as required.
WHO Three-Step Analgesic Ladder
Non-Opioid Analgesics: Paracetamol and NSAIDs
Paracetamol (acetaminophen) is the most widely used analgesic globally and the cornerstone of every step of the analgesic ladder. Despite decades of clinical use, its precise mechanism of action remains incompletely understood. It does not significantly inhibit peripheral COX enzymes at therapeutic doses. Proposed central mechanisms include inhibition of a central COX isoform (COX-3), activation of descending serotonergic inhibitory pathways, and indirect cannabinoid receptor activation via its metabolite AM404.
Pharmacokinetics: oral bioavailability approximately 80–90%; peak plasma concentration at 30–60 minutes; half-life 2–3 hours. IV paracetamol achieves faster peak CNS concentrations and is indicated when oral administration is not possible or when rapid onset is required (e.g., post-operative setting). Dose: 1 g every 4–6 hours in adults (maximum 4 g/24 h); lower doses (15 mg/kg) for children; dose reduction mandatory in severe hepatic impairment and in patients with low body weight (<50 kg): 15 mg/kg per dose (maximum 3 g/24 h). Hepatotoxicity — the key safety concern — is caused by the reactive metabolite NAPQI (N-acetyl-p-benzoquinone imine), which accumulates in overdose or in states of depleted hepatic glutathione (chronic alcoholism, malnutrition, concurrent CYP2E1-inducing drugs). At therapeutic doses, NAPQI is rapidly conjugated by glutathione and excreted harmlessly.
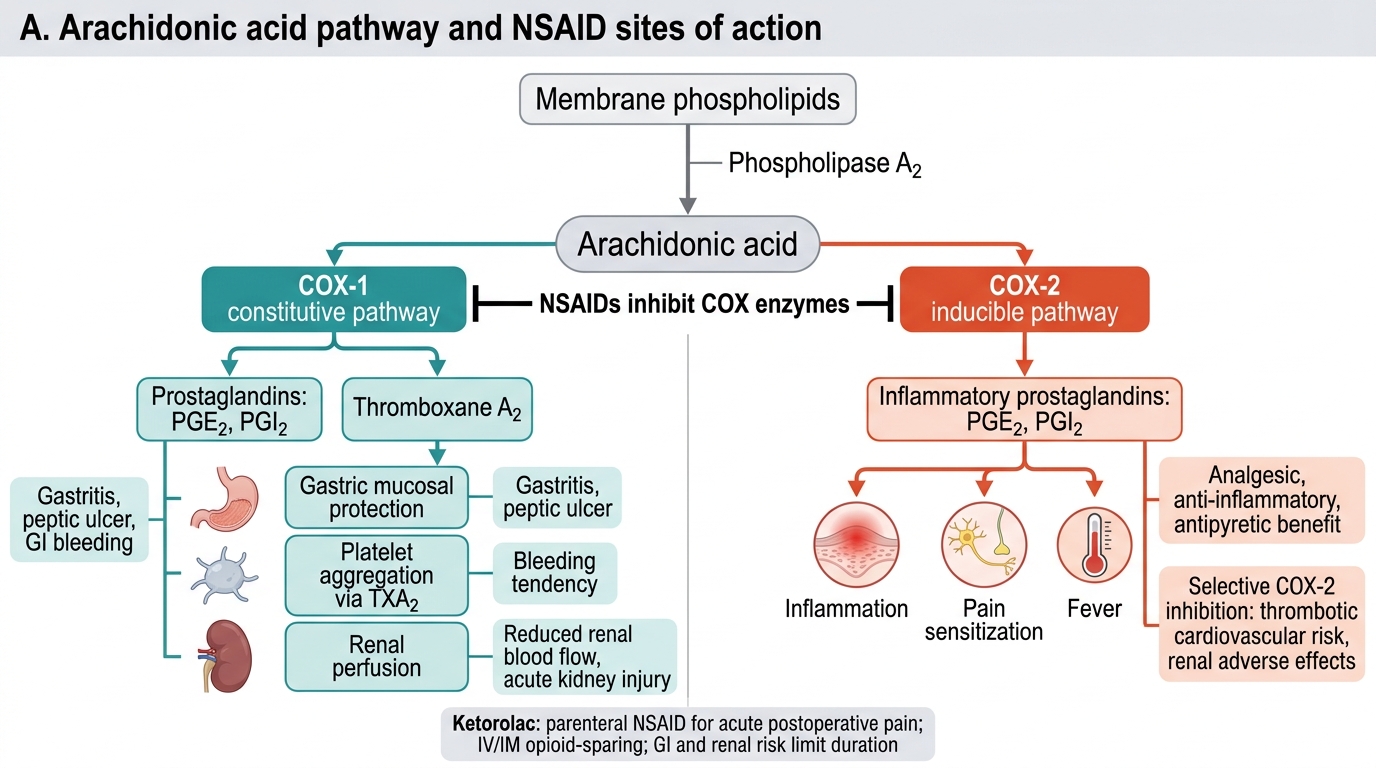
Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit COX enzymes, reducing the synthesis of prostaglandins and thromboxane A₂ from arachidonic acid. This peripheral action directly reduces nociceptor sensitisation and inflammation, providing analgesia, antipyresis, and anti-inflammatory effects. NSAIDs also have a central analgesic component through inhibition of spinal prostaglandin synthesis.
Classification by COX selectivity:
- Non-selective NSAIDs (COX-1 and COX-2 inhibitors): ibuprofen, diclofenac, naproxen, indomethacin, ketorolac. Carry risk of gastrointestinal ulceration and bleeding (COX-1 inhibition reduces protective gastric prostaglandins), platelet dysfunction (COX-1-mediated thromboxane A₂ required for aggregation — reversible for ibuprofen/diclofenac, irreversible for aspirin), and renal impairment (prostaglandin-dependent afferent arteriolar tone in states of renal hypoperfusion).
- Selective COX-2 inhibitors (coxibs): celecoxib, etoricoxib. Improved GI safety profile but carry increased cardiovascular risk (loss of prostacyclin-mediated vasodilation without proportional reduction in thromboxane A₂ → prothrombotic state).
Contraindications common to most NSAIDs: peptic ulcer disease, chronic kidney disease (eGFR <30 mL/min), significant cardiovascular disease, third trimester pregnancy (premature closure of ductus arteriosus via prostaglandin inhibition), and concurrent anticoagulation without gastroprotection.
NSAIDs and the Arachidonic Acid COX Pathway
Ketorolac deserves specific mention as the only parenteral NSAID widely available in India and globally for acute pain. Given IV or IM, it provides analgesia equivalent to 10–12 mg morphine for moderate surgical pain. Dose: 15–30 mg IV/IM (maximum 60 mg/day in adults; 5-day limit due to renal and GI risk). It is particularly valuable as an opioid-sparing agent in the immediate post-operative period.
Opioid Analgesics: Mechanisms, Classification, and Receptor Pharmacology
Opioids are the most potent analgesics available for severe nociceptive pain and remain the mainstay of Step 3 analgesia on the WHO ladder. They act on three principal receptor types — μ (mu), δ (delta), and κ (kappa) — which are G-protein-coupled receptors (GPCRs) of the Gi/o class. Activation produces: decreased adenylyl cyclase activity (reduced cAMP), decreased voltage-gated Ca²⁺ channel conductance (reduced neurotransmitter release from presynaptic terminals), and increased K⁺ channel conductance (membrane hyperpolarisation). The net effect is reduced neuronal excitability in pain-processing circuits.
Provided image
μ receptors are the primary mediators of opioid analgesia (supraspinal and spinal), euphoria, respiratory depression, physical dependence, reduced gastrointestinal motility, and miosis. Most clinical opioids are μ agonists. δ receptors modulate μ receptor function and contribute to spinal analgesia. κ receptors produce spinal analgesia and sedation but also dysphoria and psychotomimetic effects — clinically limiting their use.
Classification by efficacy:
- Full μ agonists: morphine, oxycodone, fentanyl, hydromorphone, pethidine (meperidine), methadone. No ceiling to analgesic effect — dose escalation limited only by adverse effects.
- Partial μ agonist: buprenorphine. Ceiling to respiratory depression (making it safer in overdose) but also ceiling to analgesia at very high doses. Also acts as κ antagonist.
- Mixed agonist-antagonist: pentazocine (κ agonist/μ antagonist). Limited clinical role — causes dysphoria, ceiling analgesia, and can precipitate withdrawal in opioid-dependent patients.
- Antagonists: naloxone (parenteral, short-acting, used for opioid reversal), naltrexone (oral, long-acting, used in addiction treatment).
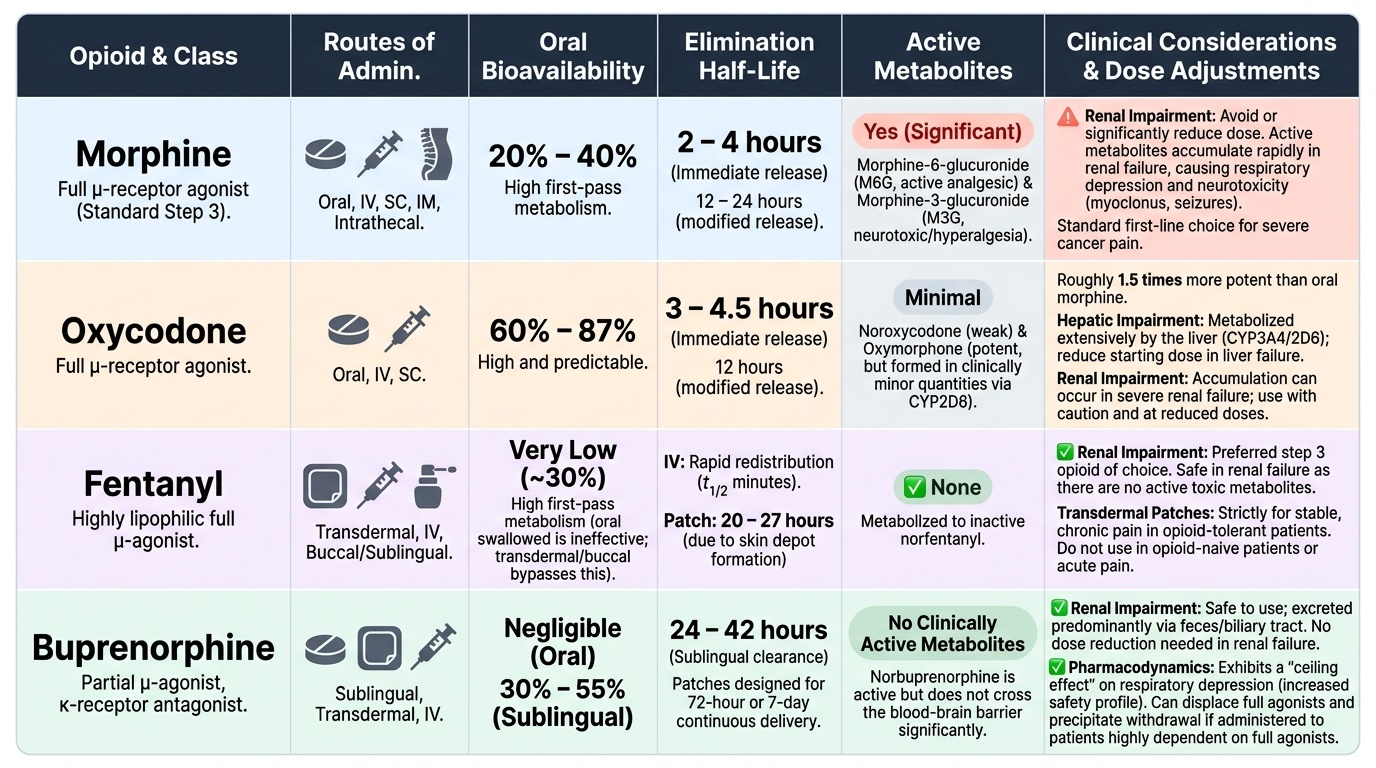
Morphine is the reference opioid and the standard against which all others are calibrated. Key pharmacology:
- Oral bioavailability: 20–30% (significant first-pass hepatic metabolism)
- Metabolised by glucuronidation to morphine-6-glucuronide (M6G) (active, 4–6× more potent than morphine, renally excreted — accumulates in renal failure causing prolonged sedation and respiratory depression) and morphine-3-glucuronide (M3G) (inactive, neuroexcitatory at high concentrations — may cause myoclonus and allodynia)
- Half-life: 2–3 hours; duration of action 4–5 hours for immediate-release formulations
- Modified-release (MR) formulations (e.g., morphine sulphate SR) provide 12-hourly dosing for stable background pain
Opioid equianalgesia is the concept of converting between opioid drugs or routes at doses producing equivalent analgesia. The reference is oral morphine. Key equianalgesic ratios (approximate, individual variation is substantial):
- Oral morphine 30 mg ≈ oral oxycodone 20 mg ≈ IV/SC morphine 10 mg ≈ IV fentanyl 100 μg
- When rotating opioids due to poor response or intolerable adverse effects, reduce the calculated equianalgesic dose of the new opioid by 25–30% to account for incomplete cross-tolerance.
- Transdermal fentanyl (patches): 25 μg/h patch ≈ oral morphine 60 mg/24 h. Take 12–24 hours to reach steady state — not suitable for acute pain titration.