Page 1 of 15
IM14.1-5 | Obesity Foundations — SDL Guide
Learning Objectives
- Define obesity using Asian-Indian BMI cut-offs (overweight ≥23, obese ≥25 kg/m²) and explain why these differ from Western thresholds
- Measure and interpret waist circumference and waist-to-hip ratio as markers of central (visceral) obesity
- Describe the aetiology of obesity: modifiable risk factors (diet, inactivity, sleep, medications), non-modifiable factors (genetics, age, ethnicity), and secondary causes (hypothyroidism, Cushing syndrome, PCOS)
- Describe the key monogenic forms of obesity (LEPR, MC4R, POMC mutations) and major syndromic forms (Prader-Willi, Bardet-Biedl)
- Apply IDF 2005 metabolic syndrome criteria with Asian-Indian waist cut-offs to clinical scenarios
- Describe the natural history of obesity and its multi-organ complications including T2DM, cardiovascular disease, NAFLD, OSA, and malignancy
INSTRUCTIONS
Obesity is the cardinal metabolic disorder of our era, and its presentation, thresholds, and management differ significantly between Indian and Western populations. This module builds your foundation: understanding how to define and measure obesity correctly in an Indian patient, recognising the full aetiological spectrum from lifestyle to rare genetic causes, applying metabolic syndrome criteria, and appreciating the multi-organ trajectory from adipose dysfunction to overt disease. Competencies IM14.1–14.5 are addressed in full.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 414 — Biology of Obesity (textbook)
- API Textbook of Medicine, 10th ed. — Obesity and Metabolic Syndrome (textbook)
- ICMR-INDIAB Study: New BMI cut-off points for defining metabolic risk in Indian adults, Journal of Association of Physicians of India 2009 (guideline)
- International Diabetes Federation: The IDF Consensus Worldwide Definition of the Metabolic Syndrome, 2005 (guideline)
- WHO Expert Consultation: Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies, Lancet 2004 (guideline)
Version 1.0 | NMC CBUC 2024
CLINICAL SCENARIO
Rajan is a 38-year-old software engineer who visits the medicine outpatient department for a routine health check. His BMI is 26 kg/m² — a number he dismisses as 'slightly overweight at most.' But when you measure his waist circumference, it is 94 cm, and his fasting glucose is 110 mg/dL. His blood pressure reads 134/86 mmHg. On examination, there are acanthosis nigricans at the neck and axillae. His mother has type 2 diabetes; his father had a myocardial infarction at 52. The Western charts Rajan looked up online told him BMI 26 is 'normal.' But for an Indian patient, a BMI of 26 kg/m² is frank obesity by the Asian-Indian definition — and his metabolic profile already shows three of the five components of metabolic syndrome. This case illustrates the most important truth about obesity in India: our thresholds differ from the West, our risks start earlier, and the metabolic consequences are already in motion long before the patient — or indeed many doctors — recognises the diagnosis.
WHY THIS MATTERS
Obesity is now the fifth leading risk factor for global mortality, directly driving type 2 diabetes, cardiovascular disease, hypertension, dyslipidaemia, non-alcoholic fatty liver disease, obstructive sleep apnoea, certain cancers, and osteoarthritis. In India, obesity prevalence has more than doubled over the past two decades; the National Family Health Survey (NFHS-5, 2019–21) reports that 24% of women and 23% of men in urban areas are overweight or obese. For the final-year MBBS student, competency IM14.1–14.5 requires not just knowing the definition but applying Indian-specific criteria, recognising secondary causes, and understanding the full trajectory of natural history and complications — because obesity will present in every discipline you practise.
RECALL
Activate your prior knowledge before diving in. Recall from biochemistry that adipose tissue is not merely an energy store but an active endocrine organ secreting adipokines — including leptin (satiety signal), adiponectin (insulin sensitiser), and resistin — as well as pro-inflammatory cytokines like tumour necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6). Recall from physiology that energy balance obeys the first law of thermodynamics: when energy intake chronically exceeds energy expenditure, the surplus is stored as triglycerides in adipocytes, leading to adipocyte hypertrophy and hyperplasia. From endocrinology, recall that insulin promotes glucose uptake and lipogenesis while suppressing lipolysis — making hyperinsulinaemia a key driver of fat deposition. From pathology, recall that visceral adiposity (fat around abdominal organs) is metabolically more harmful than subcutaneous fat, driving insulin resistance through a distinct pattern of adipokine release and direct portal delivery of free fatty acids to the liver.
Defining and Measuring Obesity in the Indian Population
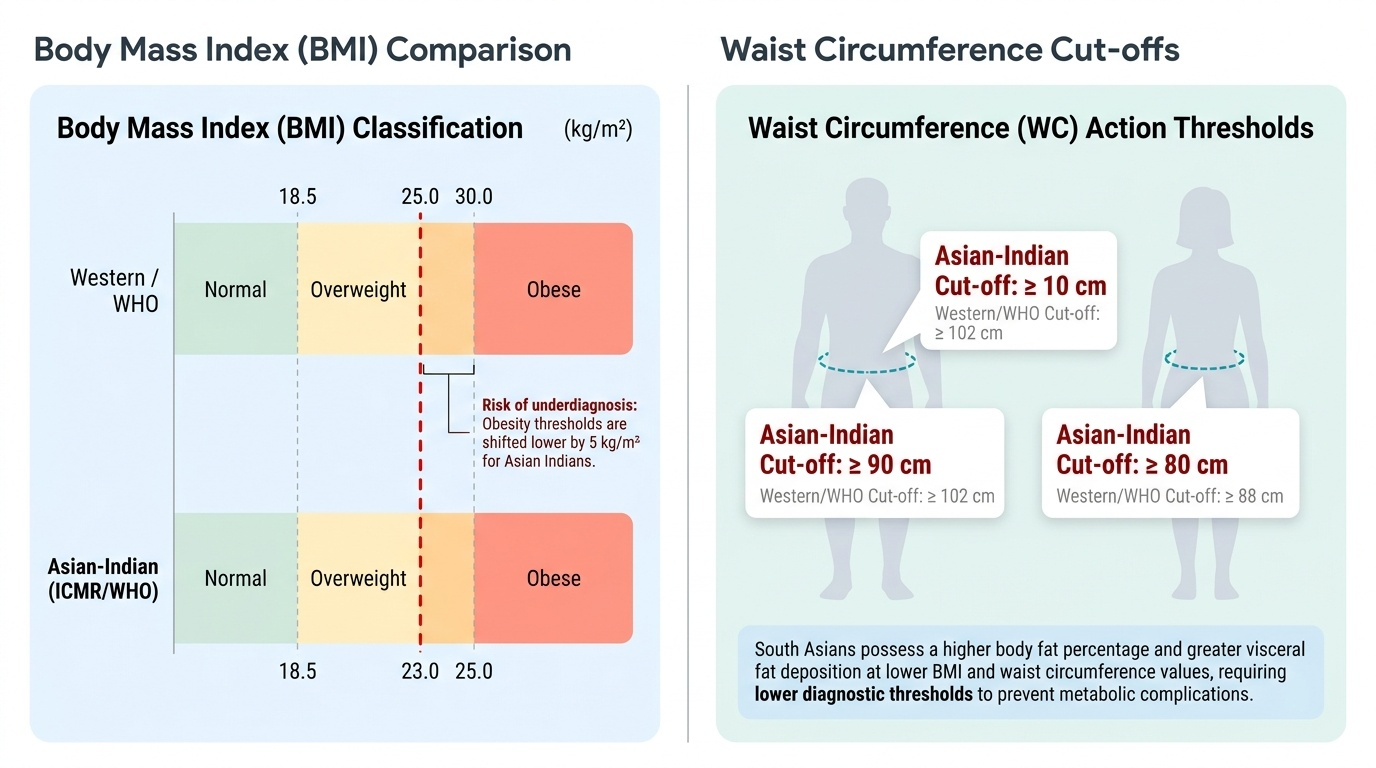
Obesity is defined as an excess accumulation of body fat to the extent that it impairs health. The conventional instrument for population-level classification is the Body Mass Index (BMI), calculated as weight in kilograms divided by the square of height in metres (kg/m²). Critically, the cut-off values that define obesity were historically derived from Western Caucasian populations. Extensive evidence over the past two decades has demonstrated that South Asians — including Indians — develop metabolic complications (insulin resistance, type 2 diabetes, hypertension, dyslipidaemia) at substantially lower BMI values than their Western counterparts, attributable to greater visceral fat deposition per unit of BMI and a higher body-fat percentage at any given BMI. These findings prompted the Indian Council of Medical Research (ICMR) and the World Health Organization (WHO) Expert Consultation to endorse ethnicity-specific cut-offs for Asian Indians.
Provided image
The Asian-Indian BMI classification is as follows:
| BMI (kg/m²) | Classification |
|---|---|
| < 18.5 | Underweight |
| 18.5 – 22.9 | Normal |
| 23.0 – 24.9 | Overweight |
| ≥ 25.0 | Obese |
Thus, overweight begins at BMI ≥ 23 kg/m² and obesity begins at BMI ≥ 25 kg/m² in Indian adults. These are not interchangeable with the Western WHO cut-offs (overweight ≥ 25, obese ≥ 30), and using the wrong thresholds will systematically underdiagnose obesity in Indian patients.
BMI alone has a well-recognised limitation: it does not capture the distribution of adiposity, and visceral (abdominal) fat is the metabolically dangerous fraction. Waist circumference (WC) is the primary clinical tool to assess abdominal (central) obesity. For Asian Indians, the action thresholds are: men ≥ 90 cm and women ≥ 80 cm. The waist-to-hip ratio (WHR) provides a complementary index; action cut-offs are WHR > 0.90 in men and > 0.85 in women. The waist-to-height ratio (WHtR) ≥ 0.5 is increasingly recognised as a strong predictor of cardiometabolic risk across ethnicities and may be even simpler to apply.
For population-level studies, skinfold thickness measurement using calipers (triceps, subscapular, biceps, suprailiac) and bioelectrical impedance analysis (BIA) are used to estimate body fat percentage. Dual-energy X-ray absorptiometry (DEXA) is the reference standard for body composition analysis in research settings. However, for routine clinical practice in India, BMI combined with waist circumference provides a practical and sufficiently accurate assessment of cardiometabolic risk.
Aetiology of Obesity: Modifiable, Non-Modifiable, and Secondary Causes
Obesity arises from a chronic positive energy balance — energy intake chronically exceeding energy expenditure over time. However, this deceptively simple equation masks a complex and interacting web of genetic, hormonal, environmental, psychological, and socioeconomic determinants that cannot be reduced to a single volitional failure. Understanding this complexity is essential for two clinical reasons: first, it allows accurate identification of remediable causes (modifiable risk factors and secondary diseases) that are often missed in busy outpatient practice; second, it provides the foundation for non-judgemental counselling that acknowledges the biological and environmental drivers of obesity rather than attributing it solely to patient willpower. For clinical and public health purposes, the aetiological framework is most usefully structured around three categories: primary multifactorial obesity (the vast majority), modifiable vs non-modifiable risk factors within that category, and secondary causes (specific pathological states causing fat accumulation, which must be excluded before labelling any patient as having lifestyle or idiopathic obesity). Each of these categories carries distinct implications for investigation, counselling, and treatment.
Non-modifiable risk factors:
- Genetic predisposition: Twin and adoption studies demonstrate that 40–70% of BMI variance is heritable. Polygenic inheritance (hundreds of small-effect single-nucleotide polymorphisms, many in or near hypothalamic appetite-regulation genes such as FTO and MC4R loci) accounts for most of the population variance.
- Age: Metabolic rate declines and lean muscle mass decreases with advancing age; fat mass increases even when intake is constant.
- Sex: Women have higher baseline body fat percentage than men; hormonal transitions (puberty, pregnancy, menopause) are inflection points for weight gain.
- Ethnicity: South Asian ancestry confers higher visceral adiposity per unit BMI independent of lifestyle, explaining the lower cut-offs.
Modifiable risk factors (environmental and behavioural):
- Dietary intake: Ultra-processed foods with high energy density, sugar-sweetened beverages, refined carbohydrates, and large portion sizes are dominant contributors. India's nutrition transition — from traditional high-fibre diets to processed foods — underlies the urban epidemic.
- Physical inactivity: Sedentary occupations, screen time, and urban design reducing walkability all contribute. The WHO recommends ≥150 minutes/week of moderate-intensity activity for adults.
- Sleep: Chronic sleep deprivation (<6 hours/night) elevates ghrelin (appetite stimulant) and suppresses leptin (satiety), driving hyperphagia.
- Stress and psychological factors: Cortisol promotes central adiposity; emotional eating is a well-recognised behavioural pattern.
- Medications as modifiable causes (iatrogenic obesity): Corticosteroids, antipsychotics (olanzapine, clozapine), antidepressants (mirtazapine, some TCAs), insulin, sulphonylureas, thiazolidinediones, valproate, lithium, and beta-blockers all promote weight gain.
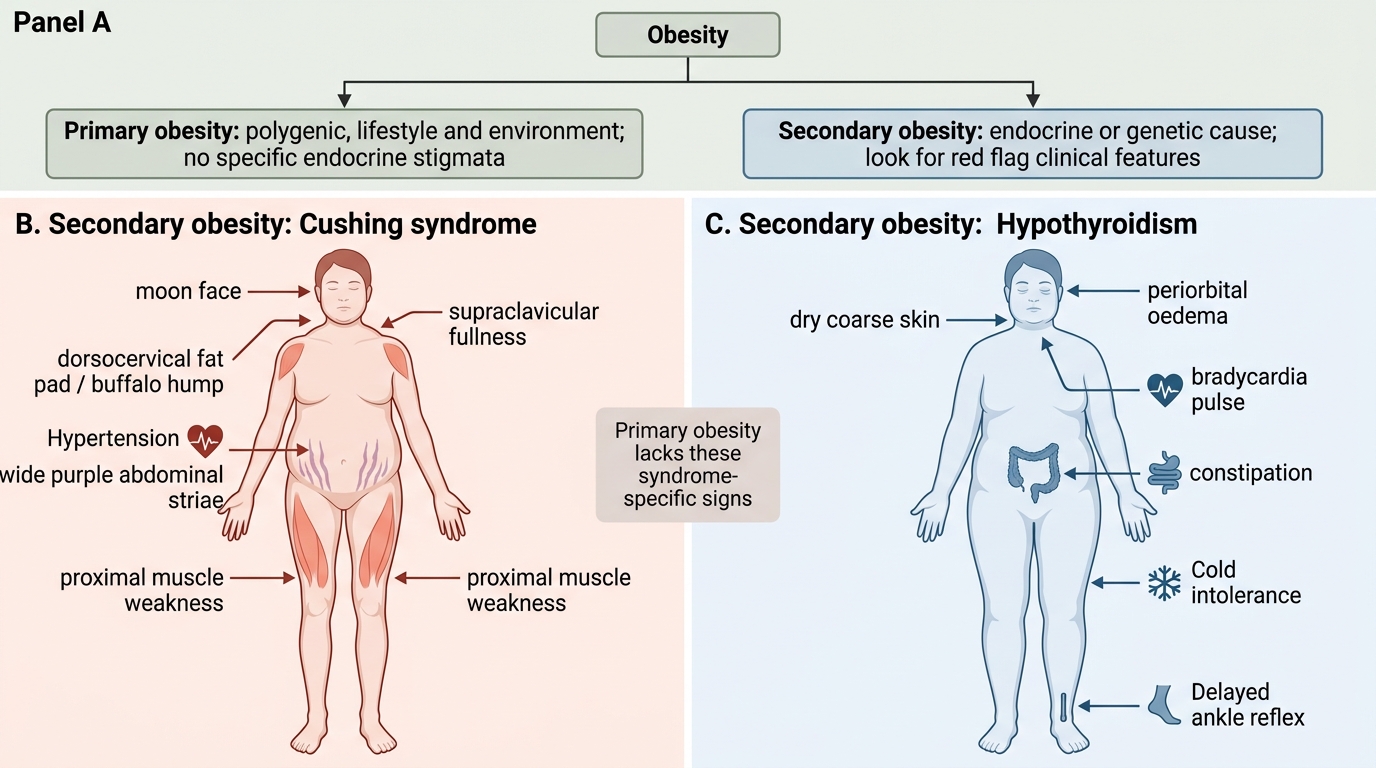
Secondary causes of obesity (must not be missed):
Secondary obesity implies a specific pathological state causing fat accumulation, often with characteristic clinical features that distinguish it from primary obesity. Always screen for these before labelling a patient as 'lifestyle obesity.'
| Secondary Cause | Key Features | Screening Test |
|---|---|---|
| Hypothyroidism | Cold intolerance, bradycardia, constipation, dry skin, periorbital oedema, elevated TSH | Serum TSH |
| Cushing syndrome | Central obesity, moon face, buffalo hump, striae (purple/wide), proximal myopathy, hypertension, hyperglycaemia | 24-h urine cortisol or 1 mg overnight dexamethasone suppression test (DST) |
| Polycystic ovary syndrome (PCOS) | Oligo/amenorrhea, hirsutism, acne, elevated androgens, polycystic ovaries on ultrasound | Testosterone, LH/FSH, pelvic ultrasound |
| Hypothalamic obesity | Headache, visual field defects, preceding trauma/surgery/radiotherapy, hyperphagia with hypodipsia | MRI brain/pituitary |
| Growth hormone deficiency | Increased body fat, reduced lean mass, fatigue, in adults (post-pituitary surgery or childhood GHD) | IGF-1, stimulation tests |
| Insulinoma | Episodic hypoglycaemia, fasting symptoms, hyperphagia, weight gain | Supervised 72-h fast, C-peptide |
Clinical Clues to Primary versus Secondary Obesity
Monogenic and Syndromic Forms of Obesity
While the vast majority of human obesity (>95%) is polygenic and multifactorial, a small but important minority is caused by single-gene mutations affecting hypothalamic appetite regulation pathways. These monogenic forms are characterised by early onset (typically in infancy or childhood), severe obesity disproportionate to the family environment, and frequently by associated endocrine or developmental abnormalities. Recognising them matters clinically because specific pharmacological targets (e.g., melanocortin-4 receptor agonists) are emerging for some.
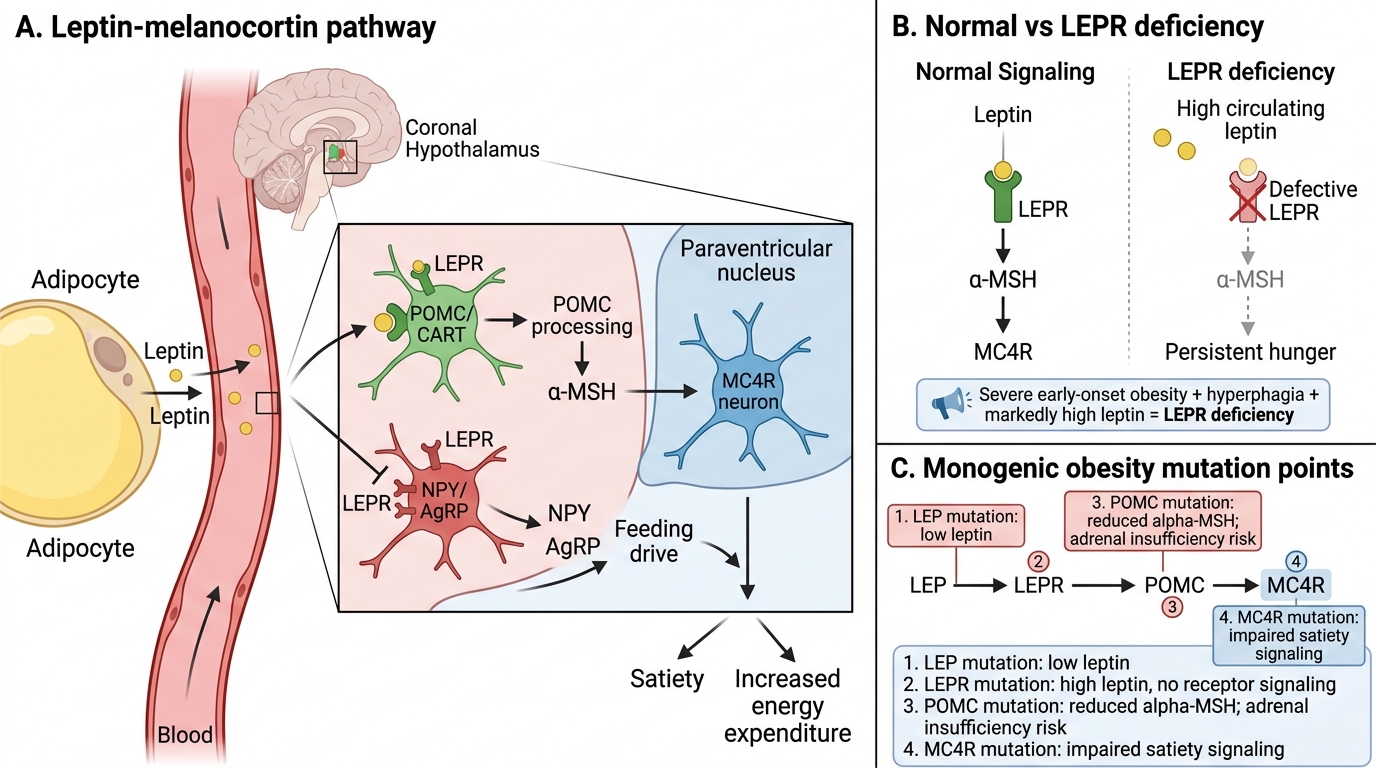
The leptin-melanocortin pathway in the hypothalamus is the primary circuit regulating long-term energy balance. Leptin, secreted by adipocytes in proportion to fat mass, acts on hypothalamic neurons expressing the leptin receptor (LEPR). Leptin binding activates proopiomelanocortin (POMC) neurons, generating alpha-MSH that binds the melanocortin-4 receptor (MC4R), producing satiety signals and increasing energy expenditure. Simultaneously, leptin suppresses neuropeptide Y (NPY) and agouti-related peptide (AgRP) orexigenic neurons. A defect at any point in this pathway abolishes satiety signalling and produces profound obesity.
Important monogenic causes in clinical practice include:
- Leptin deficiency (LEP mutation): Extremely rare; autosomal recessive. Presents with severe obesity from infancy, constant hyperphagia, low serum leptin, hypogonadotrophic hypogonadism, and immune dysfunction. Treatable with daily recombinant leptin injections — one of the few monogenic obesity syndromes with specific therapy.
- Leptin receptor deficiency (LEPR mutation): Similar phenotype to leptin deficiency but serum leptin is markedly elevated (receptor non-functional). Also autosomal recessive.
- POMC deficiency: Autosomal recessive; obesity from infancy, adrenal insufficiency (ACTH is derived from POMC), and, in some variants, red hair (due to loss of alpha-MSH stimulation of skin melanocortin receptors). Setmelanotide (an MC4R agonist) is approved for POMC-deficiency obesity.
- MC4R mutation: The most common monogenic form, accounting for 2–5% of severe childhood obesity; autosomal dominant with variable penetrance. Hyperphagia from childhood, normal or tall stature, hyperinsulinaemia. Setmelanotide also approved.
- SIM1 haploinsufficiency: Causes hypothalamic obesity; SIM1 is a transcription factor regulating Paraventricular nucleus neurons.
Syndromic obesity refers to well-defined genetic syndromes where obesity is one component:
- Prader-Willi syndrome: Paternal chromosome 15q11-q13 deletion; characterised by neonatal hypotonia, poor feeding in infancy, then hyperphagia and rapid weight gain in childhood, intellectual disability, short stature, hypogonadism, and behavioural problems. Growth hormone therapy is used.
- Bardet-Biedl syndrome: Autosomal recessive, mutations in BBS genes (ciliopathy); obesity, retinal dystrophy (night blindness), polydactyly, renal anomalies, intellectual disability, and hypogonadism.
- Alström syndrome: Autosomal recessive (ALMS1); childhood obesity, sensorineural hearing loss, retinal degeneration, dilated cardiomyopathy.
Leptin-Melanocortin Pathway in Obesity
SELF-CHECK
A 4-year-old boy is brought for evaluation of severe obesity since infancy with constant hunger and inability to be satiated. His BMI is at the 99th centile. Serum leptin is markedly elevated. His parents and older sibling are of normal weight. What is the most likely monogenic cause?
A. Leptin (LEP) gene mutation causing leptin deficiency
B. Leptin receptor (LEPR) gene mutation causing receptor deficiency
C. Melanocortin-4 receptor (MC4R) mutation
D. Proopiomelanocortin (POMC) mutation with adrenal insufficiency
Reveal Answer
Answer: B. Leptin receptor (LEPR) gene mutation causing receptor deficiency
The hallmark of LEPR (leptin receptor) deficiency is severe early-onset obesity with constant hyperphagia AND markedly elevated serum leptin levels — because leptin is produced normally by fat tissue but cannot signal through its non-functional receptor. In contrast, LEP (leptin deficiency) would show very LOW serum leptin. MC4R mutation is autosomal dominant (family members would often be affected) and serum leptin may be elevated but not as markedly. POMC deficiency would include adrenal insufficiency and possibly red hair, not mentioned here. The combination of severe obesity + markedly elevated leptin in an isolated case points to LEPR deficiency.