Page 4 of 14
IM19.3-6 | Movement Disorder Clinical Evaluation — SDL Guide
Learning Objectives
- Elicit, document, and present a structured history for a patient with a movement disorder, including onset, progression, precipitating/aggravating/relieving factors, and associated symptoms
- Perform a systematic general and detailed neurological examination in a patient with a movement disorder, including the use of standard movement rating scales
- Generate a prioritised differential diagnosis based on history and physical examination findings
- Synthesise clinical findings to determine the anatomical location, nature, and likely cause of the movement disorder
INSTRUCTIONS
The clinical evaluation of a patient with a movement disorder is a structured skill that integrates focused history-taking, systematic physical examination, and pattern recognition into a coherent diagnostic formulation. Unlike many areas of medicine where laboratory investigations drive diagnosis, movement disorders are fundamentally diagnosed at the bedside — by how the movement looks, when it occurs, what makes it better or worse, and what accompanies it. This module guides you through each step of that clinical assessment, from the first questions in the history to the standardised rating scales used to quantify severity, culminating in the anatomical localisation and differential diagnosis that guide investigation.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 431 — Movement Disorders, Clinical Assessment (textbook)
- API Textbook of Medicine, 10th ed., Ch. — Extrapyramidal Disorders: Clinical Approach (textbook)
- Davidson's Principles & Practice of Medicine, 24th ed., Ch. 28 — Neurological Examination (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
The neurology registrar presents a patient to you during the ward round: 'This is Mr Suresh, 58 years old, referred for a movement disorder.' That is all you know. You have ten minutes to take a history and examine him. Where do you start? What questions will tell you whether this is tremor or rigidity, Parkinson's or essential tremor, functional or organic? Which examination manoeuvres will reveal bradykinesia and which will distinguish cerebellar from extrapyramidal disease? And once you have your clinical findings, how do you synthesise them into an anatomical location, a nature, and a cause? The examination room is both the diagnostic laboratory and the procedural suite for movement disorders. No blood test tells you whether the tremor is at rest or with action; no MRI reveals cogwheel rigidity. The clinician's trained hands, eyes, and ears are the primary instruments — everything else is confirmatory.
WHY THIS MATTERS
Clinical evaluation skills in movement disorders have direct, high-stakes consequences for patients. Misidentifying the phenomenology of tremor — for example, labelling essential tremor as Parkinson's disease — exposes the patient to years of unnecessary levodopa, with its own complications, while the correct and effective treatment (propranolol) remains unused. Conversely, missing bradykinesia in early Parkinson's disease delays treatment that can substantially improve quality of life. In the Indian context, where specialist neurology referral may be delayed, the general internist's ability to conduct a thorough movement disorder evaluation is often the entry point into appropriate management. The NMC IM19.3 through IM19.6 competencies are rated SH (Skills and Understanding applied to actual clinical settings) — meaning you must be able to actually perform and interpret these clinical assessments, not merely describe them. This module provides the procedural knowledge for that standard.
RECALL
Before proceeding, recall your prior learning on neurological history-taking and examination. A comprehensive neurological history uses the same structural framework as any medical history but has specific neurological dimensions: the mode of onset (sudden = vascular, trauma; subacute = inflammatory, toxic, metabolic; insidious/progressive = degenerative, neoplastic), the progression (improving = typically ischaemic or toxic; static = structural; worsening = neurodegenerative, mass), and the temporal pattern (episodic = epilepsy, migraine, TIA; continuous). You should also recall the components of the neurological examination: higher mental function, cranial nerves, motor system (tone, power, reflexes, coordination), sensory system, and gait. For this module, the motor system examination and gait are the central focus — specifically the extrapyramidal components (tone by cogwheeling, bradykinesia testing by rapid alternating movements, tremor characterisation by activation condition) that are not always covered in the basic motor examination taught in pre-clinical years.
Clinical Indication and Relevance: Why a Structured Assessment Matters
A structured clinical assessment for movement disorders is indicated whenever a patient presents with abnormal involuntary movements, motor slowness, tremor, altered gait, postural instability, or any complaint that suggests dysfunction of the motor system beyond weakness or sensory loss. The clinical evaluation is the cornerstone of movement disorder diagnosis because the phenomenology — how the movement looks, when it occurs, and what modifies it — is itself the primary diagnostic data. No imaging study can substitute for the clinician who observes that a patient's hand tremor disappears during voluntary movement (rest tremor → basal ganglia), re-emerges on sustained posture (re-emergent tremor of PD), and is accompanied by a pill-rolling quality at 4–6 Hz. These observations are available only at the bedside.
The relevance of a structured approach is threefold. First, it is efficient: a directed history that asks about onset, distribution, activation, modifying factors, and associated features narrows the differential from dozens of diagnoses to two or three before any test is ordered. Second, it is reproducible: using validated rating scales such as the MDS-Unified Parkinson's Disease Rating Scale (MDS-UPDRS) allows the severity of motor features to be quantified, compared over time, and communicated precisely between clinicians and researchers. A verbal description of 'mild tremor' is not reproducible; an MDS-UPDRS motor subscore is. Third, it is patient-centred: the history gives the patient the opportunity to describe not only the motor symptom but its functional impact — on handwriting, on daily activities, on work, on social participation — which drives treatment prioritisation.
A movement disorder evaluation should be triggered, and structured around, a set of focused clinical questions that you carry into every patient encounter. These are the questions the following sections will teach you to answer: Is the movement disorder hypokinetic or hyperkinetic? What is the exact phenomenological category (tremor type, chorea, dystonia, myoclonus, tics)? What is its distribution (focal, segmental, hemibody, generalised)? What is its onset and progression pattern? Are there associated neurological signs that localise the problem (e.g., pyramidal signs → UMN involvement, cerebellar signs → cerebellar pathway, autonomic features → MSA, cognitive features → DLB or Huntington's)? Are there systemic features that point to an aetiology (liver disease → Wilson's, cardiac → rheumatic fever/Sydenham's, drug history → drug-induced)?
Governing Principles: History-Taking in Movement Disorders
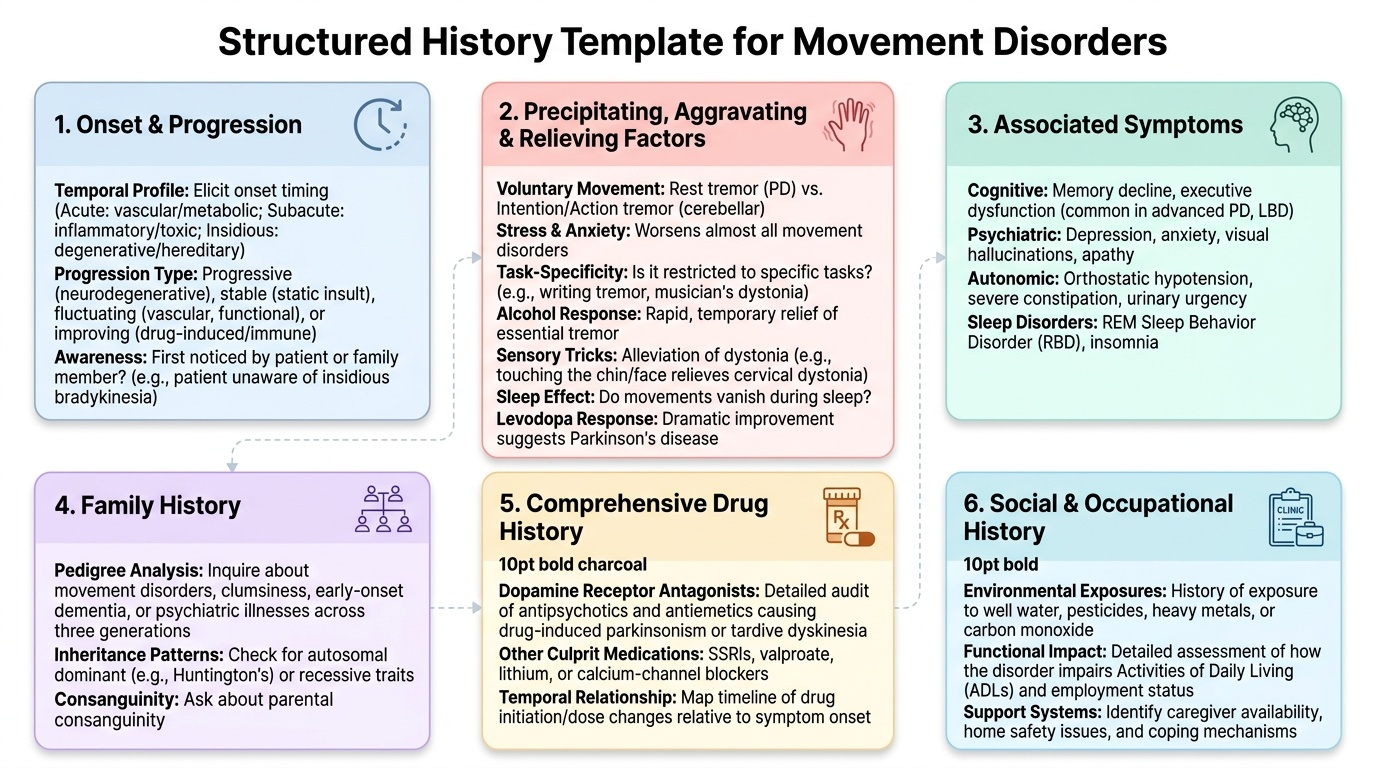
The history in movement disorders follows the standard medical framework but has specific extensions for neurological localisation and aetiological diagnosis. The following components must be elicited and documented for every patient with a movement disorder, in the order that best flows from the patient's narrative while ensuring completeness.
Provided image
Onset and progression:
Establish exactly when the movement disorder began. Was it acute/sudden (hours-days → vascular, toxic, metabolic, inflammatory), subacute (days-weeks → inflammatory, toxic, paraneoplastic), or insidious/gradual (months-years → degenerative, hereditary)? Was it first noticed by the patient or by a family member (patients with bradykinesia often do not perceive their own slowing until it is pointed out; patients with tics may have suppressed them for years)? Once started, has it worsened progressively (most neurodegenerative), stayed stable (often toxic, post-traumatic, metabolic after cause resolved), improved (Sydenham's chorea, drug-induced), or fluctuated (vascular, functional)?
Precipitating, aggravating, and relieving factors:
These are diagnostically powerful and must be asked explicitly because patients rarely volunteer them:
- Voluntary movement: rest tremor of PD disappears with movement; intention tremor of cerebellar disease worsens with movement.
- Stress and anxiety: worsens all tremors but particularly essential tremor and functional tremor; also worsens tics.
- Specific tasks: task-specific tremors (writing tremor, musician's dystonia) only present during the triggering task — the patient is tremor-free at other times.
- Alcohol: essential tremor is characteristically relieved by small amounts of alcohol (not a treatment, but a history clue); PD tremor is not reliably alcohol-responsive.
- Sensory tricks (geste antagoniste): touching the chin or face relieves cervical dystonia; this sensory trick is specific to dystonia.
- Sleep: virtually all movement disorders except palatal tremor and some epileptic myoclonus disappear during sleep.
- Levodopa response: improvement with levodopa suggests nigrostriatal dopaminergic deficiency (PD, dopamine-responsive dystonia DRD). The response itself is diagnostic — 'Is there a family history of similar problems responding dramatically to small doses of levodopa?' points to DRD (Segawa disease).
Associated symptoms:
A movement disorder does not exist in isolation; associated features localise and classify the disease:
- Cognitive decline → Huntington's, DLB, PSP
- Psychiatric symptoms (depression, anxiety, impulsivity) → Huntington's, PD, Wilson's
- Autonomic features (orthostatic hypotension, urinary dysfunction, erectile dysfunction, constipation) → PD, MSA
- Sleep disturbance (REM sleep behaviour disorder — acting out dreams, kicking and shouting during sleep) → early marker of PD, MSA, DLB
- Falls early in the course → PSP (early postural instability and vertical gaze palsy), MSA
- Dysphagia/dysarthria early → PSP, MSA
- Visual hallucinations → DLB
- Sore throat or streptococcal illness → Sydenham's chorea (acute rheumatic fever)
- Liver disease, psychiatric symptoms in a young person → Wilson's disease
- Drug history → antipsychotics, metoclopramide, domperidone, sodium valproate, lithium, phenytoin, amiodarone, calcium channel blockers (flunarizine, cinnarizine) — all can cause drug-induced movement disorders
Family history:
Autosomal dominant: essential tremor, Huntington's disease, DYT1 dystonia (some forms), spinocerebellar ataxias. Autosomal recessive: Wilson's disease (ATP7B), DYT1 generalized torsion dystonia (non-manifesting carrier parents), Friedreich's ataxia. Maternal inheritance: mitochondrial disorders.
Social history:
Occupation (exposure to toxins — manganese mining → manganism, mimic parkinsonism; MPTP from illicit drug synthesis; organophosphate in agriculture); place of birth (Wilson's disease higher prevalence in India, especially in certain founder-effect regions); recreational alcohol intake (essential tremor history clue).
Procedure and Technique: Neurological Examination for Movement Disorders
The neurological examination for movement disorders begins with general observation and proceeds systematically through tone assessment, motor testing, gait analysis, and specific manoeuvres for each phenomenological category. The examination is as much about what you observe spontaneously as what you elicit by specific tests — a trained examiner begins characterising the movement disorder from the moment the patient walks through the door.
General observation (before formal examination):
Observe the patient walking in from the waiting room or entering the consulting room. Note: is the gait slow and shuffling with reduced arm swing (parkinsonian)? Is there a stooped, flexed posture? Does the patient make involuntary dance-like movements while trying to sit still (chorea)? Are there sustained abnormal postures (dystonia)? How does the patient undress and dress — are the fine finger movements slow and clumsy? Is there mask-like facies (hypomimia — reduced facial expression, a feature of PD)? Observe spontaneous blinking rate (reduced in PD — normal 15–20 blinks/min, PD often <10).
Assessment of tone:
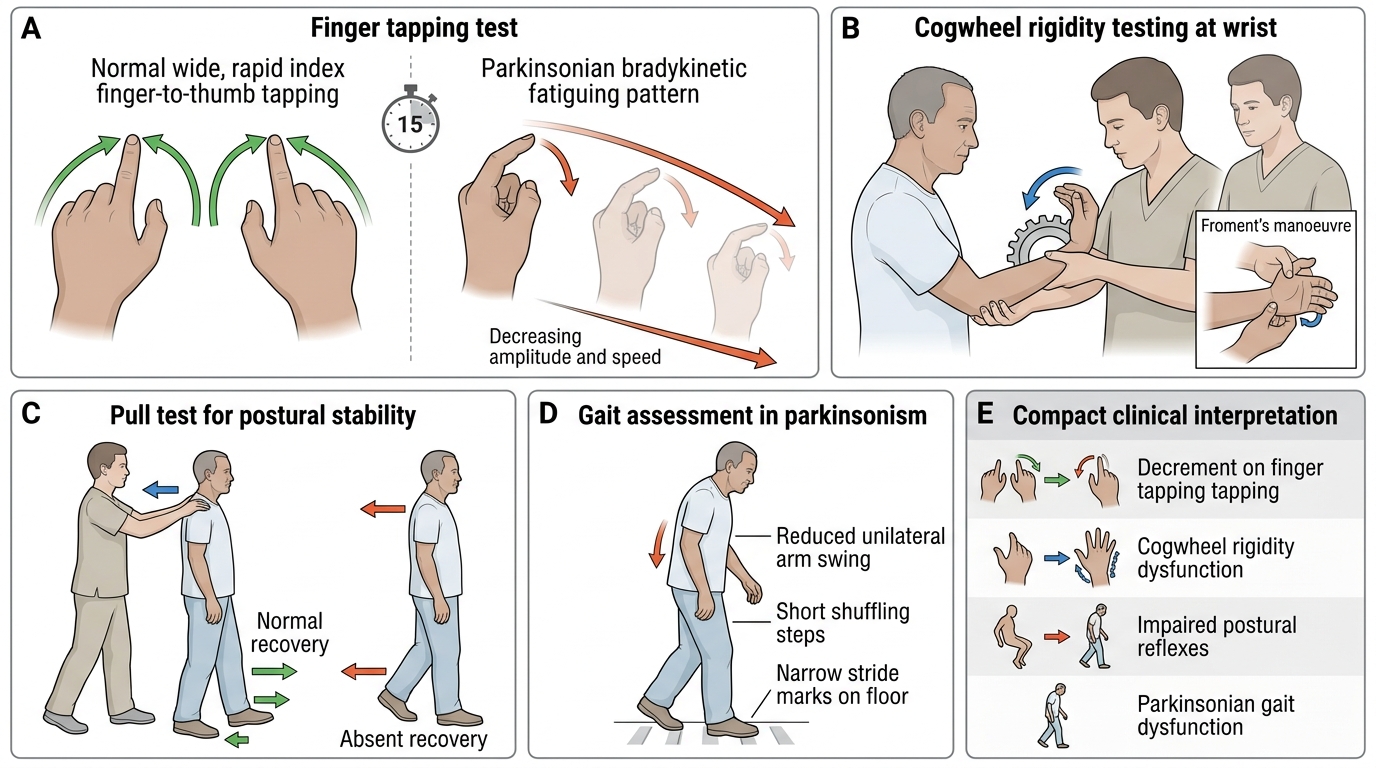
Tone is assessed by passive movement of the limb through its range of motion at the wrist, elbow, knee, and ankle. In normal subjects, there is minimal resistance. Lead-pipe rigidity is uniform, velocity-independent resistance throughout the range of motion — palpable and visible as a smooth resistance to passive flexion-extension or rotation. Cogwheel rigidity is lead-pipe rigidity with superimposed tremor — producing a ratchet-like series of catch-and-release sensations. To sensitise the test for subtle rigidity, ask the patient to perform a voluntary motor task with the contralateral limb (e.g., 'open and close your other hand repeatedly') while you passively rotate the ipsilateral wrist — this Froment's manoeuvre brings out subtle cogwheeling that may be missed on standard examination. Distinguish from spasticity (UMN): spasticity is velocity-dependent (resistance increases with faster passive movement — the 'clasp-knife' phenomenon at speed), while rigidity is velocity-independent.
Assessment of bradykinesia:
Bradykinesia is slowed, reduced-amplitude voluntary movement, typically worsening with repetition (fatiguing). The standard bedside tests for bradykinesia correspond directly to MDS-UPDRS Item 3 (Motor Examination):
1. Finger tapping (MDS-UPDRS 3.4): ask the patient to tap the index finger against the thumb as rapidly and as widely as possible. Observe speed, amplitude, and fatiguing (does the movement get progressively smaller and slower after 10–15 taps? — this is highly specific for bradykinesia).
2. Hand movements (MDS-UPDRS 3.5): opening and closing of the hand rapidly and fully. Same criteria for fatiguing.
3. Rapid alternating hand movements (pronation-supination) (MDS-UPDRS 3.6): ask the patient to alternately pronate and supinate the hand on the thigh as rapidly as possible. Slowed, irregular, or small-amplitude movements suggest bradykinesia; this also tests for dysdiadochokinesia (cerebellar — irregular, not fatiguing, with equal amplitude throughout).
4. Foot tapping (MDS-UPDRS 3.7): tapping the heel on the floor or the toe on the ground as fast and high as possible. Lower limb bradykinesia may be out of proportion to upper limb.
5. Leg agility (MDS-UPDRS 3.8): rapidly lifting and stamping the leg up and down.
IMPORTANT: the MDS-UPDRS motor section (Part 3) is the standard validated instrument for quantifying motor features of parkinsonism. Score each item 0–4 (0=normal, 4=severe). The total MDS-UPDRS Part 3 score correlates with disease severity and is used in clinical trials and monitoring. Each motor item is scored both for the right and left sides, capturing asymmetry — a feature that supports idiopathic PD (asymmetric at onset) vs drug-induced parkinsonism (usually symmetric) vs Parkinson-plus (variably symmetric).
Tremor characterisation:
Once a tremor is observed, characterise it systematically:
1. At rest: ask the patient to relax the arms fully on the lap or armrests — does tremor emerge at rest? Note its frequency (count oscillations/second if possible or estimate) and amplitude. Does it disappear when the patient is distracted or asked to perform a mental task? (Some functional tremors diminish dramatically with distractibility — see Hoover's test for functional disease.)
2. With posture: ask the patient to hold the arms outstretched horizontally for 15–30 seconds — does tremor appear or increase? If there was a rest tremor, does it disappear transiently and then re-emerge at posture (re-emergent tremor — characteristic of PD, typically occurs after a short latency of 2–10 seconds, has the same frequency as the rest tremor)?
3. With intention (kinetic): ask the patient to perform finger-nose testing — does the tremor worsen progressively as the finger approaches the nose? Note dysmetria and past-pointing as additional cerebellar signs.
Gait examination:
Ask the patient to walk 10 metres, turn, and walk back. Observe:
- Stride length and speed (reduced in parkinsonism — shuffling gait)
- Arm swing (reduced, absent, or asymmetric in PD — arm swing asymmetry is a sensitive early sign)
- Posture (flexed, stooped, head and trunk anteropulsion in PD; retropulsion and erect posture in PSP)
- Initiation (difficulty starting to walk — start hesitation or festination)
- Turns: PD patients turn en bloc (multiple small steps) rather than in one smooth motion
- Freezing episodes: sudden inability to continue walking, as if feet are stuck to the floor (freezing of gait) — a feature of advanced PD and PSP
- Romberg's test: standing with feet together, eyes open and closed — wobble or fall suggests sensory ataxia or cerebellar disease rather than extrapyramidal disease
- Tandem walking: walk heel-to-toe in a straight line — sensitive for cerebellar ataxia and impaired balance
Postural stability (pull test):
Stand behind the patient with the patient's feet shoulder-width apart. Warn them you will pull backwards. Give a brisk pull on the shoulders and observe the recovery response. Normal: one or two recovery steps. Abnormal (impaired postural reflexes): multiple steps, retropulsion, or falling without attempting to step (PIGD phenotype in PD). This corresponds to MDS-UPDRS 3.12 (postural stability). Impaired postural stability early in the disease course suggests Parkinson-plus rather than idiopathic PD.
Examinations specific to other movement disorders:
- Chorea: irregular, random movements observable during the general inspection; ask patient to stick tongue out and hold it still (chorea prevents sustained tongue protrusion — serpentine tongue in Huntington's); assess for milkmaid grip (irregular squeezing when the patient grips the examiner's two fingers).
- Dystonia: look for sustained abnormal postures at rest and during voluntary movement; apply the sensory trick (touch patient's chin/face for cervical dystonia); score using the Burke-Fahn-Marsden Dystonia Rating Scale for generalised dystonia.
- Tics: observe for stereotyped, suppressible movements or vocalisations; ask about premonitory urge and ability to suppress.
Bedside Examination for Parkinsonism
SELF-CHECK
During a bedside examination of a 60-year-old man with suspected parkinsonism, you ask him to tap his right index finger against his right thumb as rapidly and as widely as possible for 15 seconds. Initially the movements are present but over 10–15 repetitions they become progressively smaller and slower. What does this finding specifically indicate and what is this phenomenon called?
A. Spasticity — the patient has an upper motor neuron lesion causing velocity-dependent increased tone
B. Dysmetria — the patient has a cerebellar hemisphere lesion on the right
C. Bradykinesia with fatiguing — consistent with dopaminergic nigrostriatal pathway dysfunction
D. Functional movement disorder — the patient is voluntarily slowing down
Reveal Answer
Answer: C. Bradykinesia with fatiguing — consistent with dopaminergic nigrostriatal pathway dysfunction
Progressive decrease in speed and amplitude of repetitive voluntary movements on sustained performance is called fatiguing or decrement — the hallmark of bradykinesia in basal ganglia disease (particularly PD). This is tested in MDS-UPDRS Part 3 Item 3.4 (finger tapping). Spasticity is a tone abnormality (velocity-dependent resistance to passive movement), not a test of voluntary movement speed. Cerebellar dysmetria produces irregularity and overshoot but not a systematic decrement in amplitude with repetition. Functional disorders may show inconsistency or improvement with distraction, not a uniform progressive decrement.