Page 17 of 24
IM2.18 | Dyslipidemia Management in IHD — SDL Guide
Learning Objectives
- Classify dyslipidaemias and describe the clinical presentations of lipid disorders
- Explain the pathogenesis of atherogenic dyslipidaemia and its role in IHD
- Interpret a lipid profile and identify treatment targets based on cardiovascular risk category
- Discuss the indications, formulations, doses, side effects, and monitoring requirements for drugs used in dyslipidaemia — statins, ezetimibe, fibrates, PCSK9 inhibitors, and omega-3 fatty acids
INSTRUCTIONS
Dyslipidaemia management is the pharmacological cornerstone of IHD prevention and secondary prevention. This module builds the clinical framework for classifying, investigating, and treating lipid disorders — with emphasis on correct drug selection, dose titration, side-effect monitoring, and the evidence base underpinning each treatment decision.
References
- Harrison's Principles of Internal Medicine, 21st ed., Ch. 407 — Disorders of Lipoprotein Metabolism (textbook)
- API Textbook of Medicine, 10th ed., Ch. on Dyslipidaemias (textbook)
- ESC/EAS Guidelines for the Management of Dyslipidaemias 2019 (guideline)
- ACC/AHA 2018 Guideline on the Management of Blood Cholesterol (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Ramesh — the asymptomatic 48-year-old from module one in this cluster — returns for his lipid review six weeks after you started him on atorvastatin 40 mg. His repeat fasting lipid profile shows LDL-C 3.1 mmol/L (120 mg/dL), down from 4.1 mmol/L (158 mg/dL). He feels well but mentions muscle aching in his thighs that began three weeks ago. He has a family history of 'heart attacks before 50'. His cardiologist had calculated his 10-year ASCVD risk at 18% (high risk) and targeted LDL-C below 70 mg/dL. At 120 mg/dL, Ramesh has not reached his LDL target, and he has a drug side effect. What do you do next: intensify the statin, switch to a different agent, add a non-statin drug, or investigate the muscle aching first? Separately, a second patient — 35-year-old Priya — has LDL-C 8.4 mmol/L (325 mg/dL) on two separate tests, tendinous xanthomata on the Achilles tendons, and a father who died of MI at age 42. This is a very different clinical picture from Ramesh: the LDL is four times the usual atherogenic threshold and the presentation suggests a genetic disorder. These two cases illustrate the full clinical range of dyslipidaemia practice — from population-level statin titration to rare but high-impact familial hypercholesterolaemia. This module provides the clinical framework for managing both.
WHY THIS MATTERS
The NMC competency IM2.18 requires knowledge-and-higher (KH) level understanding of the indications, formulations, doses, side effects, and monitoring of dyslipidaemia drugs — meaning you must be able to prescribe the right drug at the right dose, recognise side effects, and order appropriate monitoring, not merely name the drug class. In India, dyslipidaemia is underdiagnosed and undertreated — the India Heart Watch study found that fewer than 15% of high-risk Indians with elevated LDL-C are on statin therapy, and of those who are, fewer than 30% achieve their LDL-C target. Improving the quality of lipid management by final-year students who will become junior doctors is a directly measurable population health intervention.
RECALL
Recall from the IHD Foundations module: LDL-cholesterol is the primary atherogenic lipoprotein, transported by ApoB-100 and cleared by hepatic LDL receptors (LDLR). Statins inhibit HMG-CoA reductase, the rate-limiting enzyme in hepatic cholesterol synthesis, thereby reducing intracellular cholesterol and upregulating LDLR expression. Recall the lipid fractions: LDL-C (primary atherogenic target), HDL-C (cardioprotective, reverse cholesterol transport), triglycerides (TG, hypertriglyceridaemia associated with pancreatitis and metabolic syndrome), and non-HDL-C (TC − HDL; captures all atherogenic particles). From pharmacology: fibrates activate PPARα, increasing LPL expression and reducing TG; ezetimibe inhibits intestinal cholesterol absorption via the NPC1L1 transporter.
Clinical Presentation and Classification of Dyslipidaemias
Dyslipidaemia is an abnormality of plasma lipid levels encompassing elevated LDL-C, elevated TG, low HDL-C, or combinations thereof. Most dyslipidaemias in clinical practice are asymptomatic until a cardiovascular event occurs — the 'silent' nature of elevated LDL-C is one of the most important public health challenges in cardiovascular prevention. However, some dyslipidaemias produce specific clinical signs that should be recognised and that carry diagnostic and prognostic significance. These physical findings are not incidental — they represent the end-organ manifestation of years of lipid accumulation and indicate both a severe and often genetic disorder requiring urgent treatment. Identifying them on examination changes the diagnostic approach from screening to targeted cascade testing, and the treatment approach from lifestyle modification to aggressive pharmacotherapy combined with family-based detection. Knowing each sign, what it implies biochemically, and which specific lipid disorder it points to is a core clinical examination skill in internal medicine.
Clinical signs of dyslipidaemia (when present — each indicates significant and often familial hyperlipidaemia):
- Xanthelasma: yellowish deposits of lipid-laden histiocytes in the periorbital skin; associated with elevated LDL-C but also occurs in normolipidaemic individuals; soft and flat, not tender
- Tendinous xanthomata: firm, irregular lipid deposits in tendons — characteristically the Achilles tendons, extensor tendons of the hands, and patellar tendons; pathognomonic of familial hypercholesterolaemia (FH) — essentially never seen in secondary dyslipidaemia alone; indicate severe, prolonged LDL elevation
- Xanthomata tuberosum: nodular deposits at pressure points (elbows, knees); seen in severe combined hyperlipidaemia or type III hyperlipoproteinaemia (familial dysbetalipoproteinaemia — ApoE2/E2 genotype)
- Eruptive xanthomata: small yellowish papules on the trunk and buttocks in severe hypertriglyceridaemia (TG >10 mmol/L); indicate TG-induced chylomicronaemia; associated with pancreatitis risk
- Corneal arcus: grey-white lipid deposit around the periphery of the cornea; in young patients (<45 years), suggests FH; in older patients (>60 years), may be a normal ageing phenomenon and not diagnostically significant
- Lipaemia retinalis: cream-coloured blood vessels visible on fundoscopy in severe chylomicronaemia (TG >20 mmol/L); associated with acute pancreatitis risk
- Pancreatitis: the most dangerous clinical consequence of severe hypertriglyceridaemia (TG >10–15 mmol/L); mechanism: pancreatic lipase hydrolyses TG within pancreatic capillaries, releasing free fatty acids that cause local chemical injury and acute pancreatitis
Fredrickson classification of hyperlipoproteinaemias (WHO/Fredrickson, modified) provides a phenotypic classification based on the elevated lipoprotein class:
- Type I (hyperchylomicronaemia): elevated chylomicrons; lipoprotein lipase deficiency; severe hypertriglyceridaemia; pancreatitis risk; cream-coloured plasma after refrigeration
- Type IIa (familial hypercholesterolaemia): elevated LDL-C only; LDLR mutations or PCSK9 gain-of-function mutations; most common cause of premature CAD
- Type IIb (combined hyperlipidaemia): elevated LDL-C and TG (both VLDL and LDL elevated); most common mixed pattern in clinical practice
- Type III (dysbetalipoproteinaemia): elevated IDL; ApoE2/E2 genotype; tuberous xanthomata
- Type IV (hypertriglyceridaemia): elevated VLDL; most common TG pattern; associated with insulin resistance, metabolic syndrome
- Type V (combined chylomicronaemia): elevated chylomicrons and VLDL; severe pancreatitis risk
Secondary causes of dyslipidaemia must always be excluded before diagnosing primary (genetic) dyslipidaemia, as treating the secondary cause may normalise the lipid profile without the need for pharmacotherapy:
- Elevated LDL-C: hypothyroidism (most important — always check TSH in new dyslipidaemia), nephrotic syndrome, obstructive liver disease, anorexia nervosa, drugs (glucocorticoids, cyclosporine, thiazides at high dose)
- Elevated TG: type 2 diabetes (insulin resistance increases VLDL synthesis), excessive alcohol, obesity, hypothyroidism, renal failure (reduced LPL activity), drugs (beta-blockers, thiazides, isotretinoin, HIV antiretrovirals)
- Low HDL-C: smoking, obesity, sedentary lifestyle, type 2 diabetes, anabolic steroids, beta-blockers (non-selective)
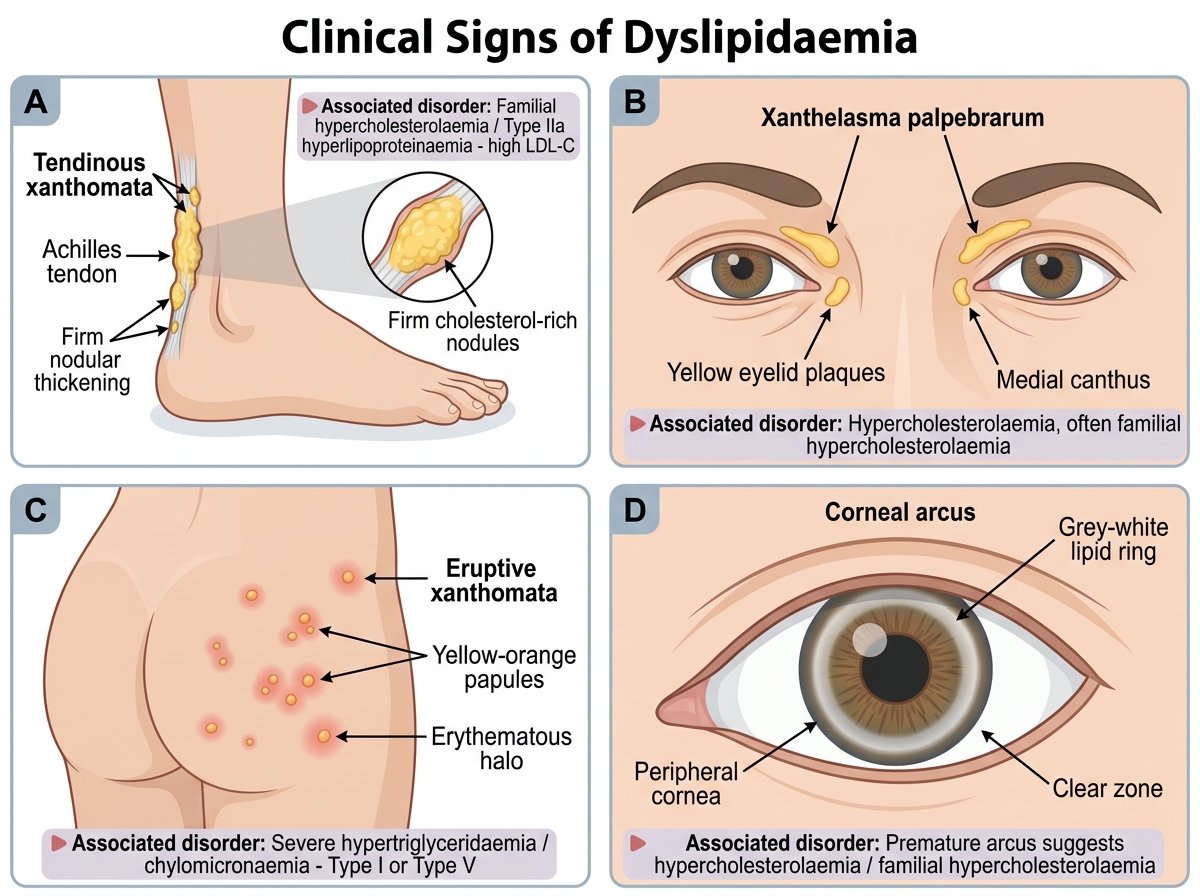
Clinical Signs of Dyslipidaemia
Pathogenesis of Atherogenic Dyslipidaemia
Atherogenic dyslipidaemia is the lipid triad of elevated LDL-C, elevated triglycerides, and low HDL-C that characterises the metabolic syndrome and type 2 diabetes mellitus — the dominant dyslipidaemia pattern in the Indian population. Understanding the pathogenesis of each component explains both the clinical risk and the rationale for each therapeutic intervention.
The central driver of atherogenic dyslipidaemia in insulin-resistant individuals is excess hepatic VLDL synthesis. In insulin resistance, adipose tissue lipolysis is incompletely suppressed by insulin, releasing excess free fatty acids (FFAs) to the liver. The liver esterifies these FFAs into triglycerides and packages them in VLDL particles. Elevated VLDL production elevates plasma TG and, via CETP (cholesteryl ester transfer protein)-mediated exchange, transfers TG into LDL and HDL in exchange for cholesterol esters. This enrichment of LDL and HDL with TG makes them better substrates for hepatic lipase, which hydrolyses the TG, producing small dense LDL (sdLDL — smaller, more atherogenic particles that penetrate the arterial intima more easily and are more susceptible to oxidation) and accelerating HDL catabolism (TG-enriched HDL is cleared faster, reducing plasma HDL-C levels). Thus, the three elements of atherogenic dyslipidaemia are biochemically linked through a single upstream mechanism — hepatic VLDL overproduction.
Familial hypercholesterolaemia (FH) has a distinct pathogenesis: mutations in the LDL receptor gene (LDLR) (heterozygous FH, incidence 1:500; homozygous FH, incidence 1:1,000,000) impair the hepatic clearance of LDL from the circulation. Without functional LDLR, LDL accumulates in plasma to 2–4× normal levels (heterozygous FH: LDL 5–10 mmol/L; homozygous FH: LDL 12–25 mmol/L). The prolonged circulating time of elevated LDL causes accelerated atherosclerosis — heterozygous FH patients have a 3–13-fold higher cardiovascular risk than the general population and develop premature CAD (men by age 50–60, women by age 60–70 without treatment). Less commonly, FH is caused by mutations in ApoB-100 (defective binding to LDLR) or PCSK9 gain-of-function mutations (PCSK9 binds LDLR and targets it for lysosomal degradation, reducing LDLR recycling and increasing plasma LDL). The Dutch Lipid Clinic Network diagnostic criteria (modified Simon Broome criteria in the UK) use family history, clinical signs, LDL-C level, and genetic testing to diagnose FH definitively.
The clinical relevance of sdLDL beyond the total LDL-C number: standard lipid assays measure LDL-C by the Friedewald equation (TC − HDL − TG/5) or by direct assay, but do not distinguish large buoyant from small dense LDL. Two patients with the same LDL-C may have very different particle numbers: the patient with atherogenic dyslipidaemia has many more sdLDL particles per unit LDL-C than the patient with simple hypercholesterolaemia. ApoB measurement directly quantifies the total atherogenic particle count (since each LDL, VLDL, IDL, and Lp(a) particle carries exactly one ApoB-100 molecule) and is a superior predictor of cardiovascular risk compared to LDL-C in patients with atherogenic dyslipidaemia.
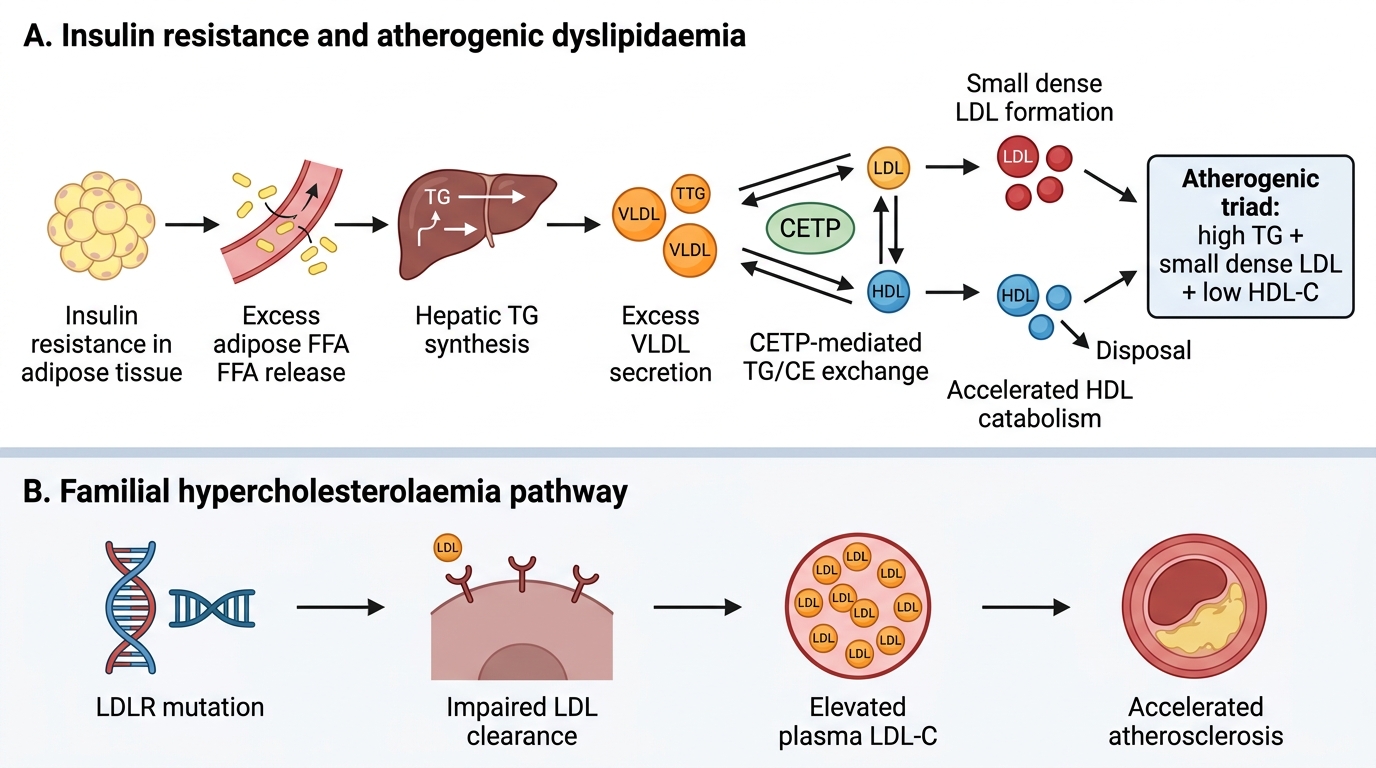
Insulin Resistance, Familial Hypercholesterolaemia, and Atherosclerosis
SELF-CHECK
A 35-year-old man has LDL-C 7.8 mmol/L (300 mg/dL) on two fasting measurements. He has tendinous xanthomata at the Achilles tendons and a family history of MI in his father at age 44. His TSH and kidney function are normal. What is the most likely diagnosis and which genetic defect is MOST commonly responsible?
A. Secondary hypercholesterolaemia from hypothyroidism — recheck TSH after optimising thyroid replacement
B. Heterozygous familial hypercholesterolaemia — most commonly caused by LDL receptor gene mutations
C. Type III hyperlipoproteinaemia (dysbetalipoproteinaemia) — ApoE2/E2 genotype
D. Mixed hyperlipidaemia (Type IIb) — elevated LDL-C and VLDL from metabolic syndrome
Reveal Answer
Answer: B. Heterozygous familial hypercholesterolaemia — most commonly caused by LDL receptor gene mutations
The clinical triad of severely elevated LDL-C (7.8 mmol/L), tendinous xanthomata (pathognomonic of FH — not seen in secondary dyslipidaemia), and premature family history of MI is diagnostic of heterozygous familial hypercholesterolaemia (FH). TSH and renal function are normal, excluding the most important secondary causes. FH is most commonly caused by mutations in the LDL receptor gene (LDLR) — over 3,000 pathogenic variants are described. Defective ApoB-100 and PCSK9 gain-of-function mutations are less common causes of the FH phenotype. Type III hyperlipoproteinaemia is associated with xanthomata tuberosum (at pressure points), not tendinous xanthomata, and with elevated IDL rather than pure LDL elevation.
Diagnosis: Lipid Profile Interpretation and Treatment Target Setting
Interpreting a lipid profile in clinical context requires more than comparing numbers against reference ranges — it requires determining the patient's cardiovascular risk category, setting individualised LDL-C targets, and deciding whether the current lipid pattern warrants pharmacological intervention or lifestyle modification alone. The same LDL-C value of 3.0 mmol/L (116 mg/dL) is below the treatment threshold in a young low-risk patient and markedly above target in a post-MI patient. This concept — that the 'normal' range for LDL-C is risk-stratified, not universal — is the most important shift in modern lipid management thinking, and understanding it prevents both under-treatment of high-risk patients (who benefit from aggressive LDL reduction) and over-medicalisation of genuinely low-risk individuals. The two major international guidelines (ESC/EAS 2019 and ACC/AHA 2018) define risk categories differently but converge on the same fundamental principle: treat the patient's risk, not just the number.
Risk-based LDL-C targets (ESC/EAS 2019 guidelines — the reference for high-risk practice in India):
- Very high cardiovascular risk: 10-year ASCVD risk >10%, OR established CVD (prior MI, ACS, PCI, CABG, TIA/stroke, peripheral arterial disease), OR type 2 diabetes with target organ damage/risk factors, OR familial hypercholesterolaemia, OR CKD stage 3–5
→ LDL-C target: <55 mg/dL (1.4 mmol/L) AND ≥50% reduction from untreated baseline
- High cardiovascular risk: 10-year ASCVD risk 5–10%, OR markedly elevated single risk factor (LDL-C >4.9 mmol/L, blood pressure ≥180/110), OR type 2 diabetes without complications
→ LDL-C target: <70 mg/dL (1.8 mmol/L) AND ≥50% reduction from baseline
- Moderate cardiovascular risk: 10-year ASCVD 1–5%
→ LDL-C target: <100 mg/dL (2.6 mmol/L)
- Low cardiovascular risk: 10-year ASCVD <1%
→ LDL-C target: <116 mg/dL (3.0 mmol/L)
ACC/AHA 2018 guidelines use a different threshold system but the core principle is the same: the higher the baseline risk, the lower the LDL-C target. The ACC/AHA uses a 10-year ASCVD risk of 7.5% (intermediate risk) and 20% (high risk) as key thresholds, with treatment intensity matched to risk tier. The most clinically important difference between ESC/EAS and ACC/AHA targets in post-ACS patients: ESC/EAS specifies <55 mg/dL; ACC/AHA specifies <70 mg/dL (both guideline-endorsed in Indian practice — some centres use ESC, others ACC/AHA; always clarify which your institution follows).
Non-HDL-C targets (= total cholesterol − HDL-C): applicable when TG is elevated (>400 mg/dL makes Friedewald-calculated LDL unreliable). Non-HDL-C target = LDL-C target + 30 mg/dL (accounts for VLDL cholesterol burden).
When to test a lipid profile:
- Screening: all adults ≥35 years (earlier if risk factors present); consider first lipid profile at age 20 in individuals with family history of premature CVD or known FH
- In ACS: at admission (within 24 hours — most accurate reflection of pre-event baseline; LDL falls after 48 hours due to acute-phase reaction)
- Monitoring on therapy: 4–12 weeks after initiating or changing lipid-lowering therapy; then annually when stable and at target
- Fasting vs non-fasting: fasting (12 hours, water permitted) is preferred for accurate TG and LDL calculation; non-fasting is acceptable for cardiovascular risk screening using non-HDL-C and total cholesterol
Cascade screening for FH: if a proband is diagnosed with FH, first-degree relatives (parents, siblings, children) should be screened for FH. Early detection and treatment initiation in FH children (before significant plaque accumulation) substantially improves long-term outcomes.
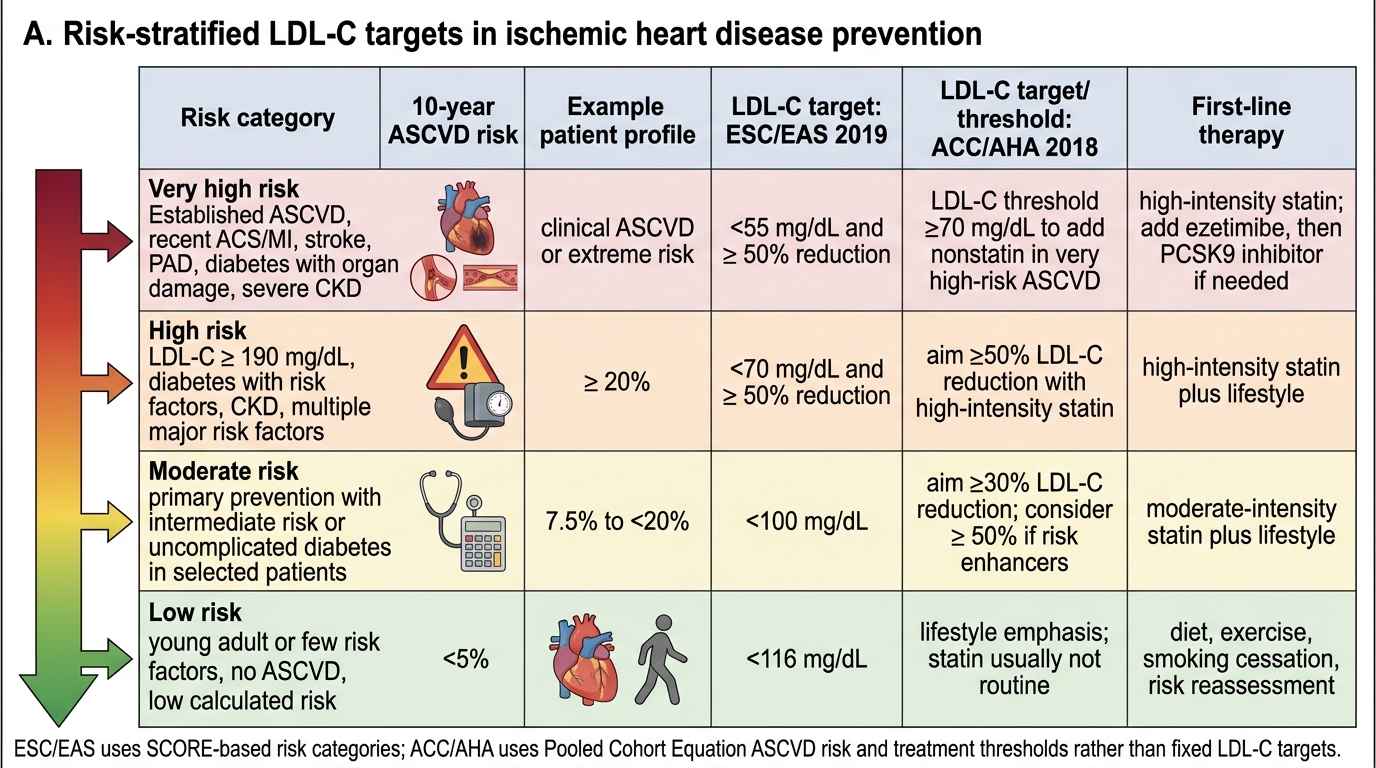
Risk-Stratified LDL-C Targets