Page 1 of 19
IM5.1-7 | Liver Disease Foundations — SDL Guide
Learning Objectives
- Describe the physiological and biochemical basis of hyperbilirubinaemia, distinguishing pre-hepatic, hepatic, intrahepatic cholestatic, and extrahepatic cholestatic causes
- Explain the epidemiology, transmission, immunology, and clinical evolution of hepatitis viruses A, B, C, D, and E
- Describe the pathophysiology of alcoholic liver disease from steatosis through alcoholic hepatitis to cirrhosis, including the role of acetaldehyde and the AST:ALT ratio
- Apply the Child-Pugh and MELD scoring systems correctly, identifying the five parameters of Child-Pugh and the three laboratory values of MELD without cross-contamination
- Enumerate the major complications of cirrhosis and portal hypertension — variceal haemorrhage, ascites (SAAG), SBP (PMN threshold), hepatic encephalopathy (West-Haven grades), HRS, and HCC — and their pathophysiological basis
- Distinguish intrinsic from idiosyncratic DILI, describing the paracetamol/NAPQI mechanism and the role of N-acetylcysteine
- Describe the pathophysiology of cholelithiasis (cholesterol vs pigment stones) and the complications of gallstone disease including biliary colic, cholecystitis, choledocholithiasis, and Charcot's triad
INSTRUCTIONS
This module establishes the pathophysiological foundations for all clinical aspects of liver disease covered in subsequent modules. Master the mechanisms first — bilirubin biochemistry, the viral hepatitis spectrum, and the cirrhosis complication cascade — because every diagnostic and management decision in hepatology flows from this framework.
References
- Harrison's Principles of Internal Medicine, 21st ed., Chapters 329–340 — Liver Disease (textbook)
- Davidson's Principles and Practice of Medicine, 23rd ed., Ch. 22 — Liver and Biliary Tract Disease (textbook)
- API Textbook of Medicine, 10th ed., Section on Hepatology (textbook)
- AASLD/EASL Guidance on Management of Cirrhosis, 2021 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
Rahul is 38 years old when he presents to the medical outpatient clinic with a three-week history of yellow eyes, swelling of both legs, and a progressively distended abdomen. His wife mentions he has been drinking heavily for nearly a decade. On examination, his sclera are deeply icteric, his abdomen is massively distended with a fluid thrill, his palms are erythematous, and there are prominent dilated veins running across his abdomen — the caput medusae. His serum bilirubin is 6.8 mg/dL, albumin is 2.1 g/dL, INR is 2.4, and he has asterixis on flapping tremor testing. Now contrast this with Priya, a 25-year-old nursing student who returns from a rural posting feeling unwell with jaundice, nausea, and right upper quadrant discomfort. Her ALT is 1,840 IU/L. She had no prior liver disease and gave a vaccination history that omitted hepatitis A. Both patients are jaundiced, but their liver diseases are entirely different in mechanism, natural history, and management. Understanding liver disease means mapping from symptom to aetiology, from aetiology to pathophysiology, and from pathophysiology to complications — the framework this module provides.
WHY THIS MATTERS
Liver disease is one of the highest-burden gastrointestinal conditions in India. Viral hepatitis B and C together affect approximately 60 million Indians; alcohol-related liver disease is the leading cause of cirrhosis in tertiary hepatology centres; and non-alcoholic fatty liver disease (NAFLD/NASH) is rapidly emerging alongside the type 2 diabetes and obesity epidemic. As a final-year student and future clinician, you will encounter jaundice, ascites, and hepatic encephalopathy on general medical wards, in emergency departments, and in outpatient clinics. The ability to identify the stage of liver disease, recognise life-threatening complications, and institute time-sensitive management is a core clinical competency at the NMC KH (Knowledge and Understanding) level — meaning you must be able to apply this knowledge to patient scenarios, not merely recall definitions.
RECALL
Before proceeding, activate your prior knowledge of normal hepatic physiology. The liver is the central metabolic organ, performing bilirubin conjugation and excretion, synthesis of plasma proteins (albumin, clotting factors, complement), gluconeogenesis and glycogen storage, fatty acid metabolism and lipoprotein assembly, bile acid production, and first-pass drug metabolism via cytochrome P450 enzymes. Bilirubin is the breakdown product of haem from senescent red blood cells: unconjugated (indirect) bilirubin is water-insoluble and transported to the liver bound to albumin; the liver conjugates it to bilirubin glucuronide (direct bilirubin) via UDP-glucuronosyltransferase (UGT1A1), making it water-soluble for excretion into bile. The portal vein drains the gut and spleen into the liver, delivering absorbed nutrients and metabolised gut bacteria products. Normal portal venous pressure is 5–10 mmHg; values above 12 mmHg define portal hypertension and drive the complications of cirrhosis. Recall also that the hepatic acinus has zones 1 (periportal, well-oxygenated, affected first in ischaemia) and zone 3 (centrilobular, most vulnerable to toxins and hypoxia in congestive or drug injury).
Bilirubin Metabolism and Hyperbilirubinaemia
Hyperbilirubinaemia — the biochemical basis of jaundice — is caused by an imbalance between bilirubin production and elimination. Understanding which step in bilirubin metabolism is disrupted allows the clinician to localise the cause before any imaging is ordered, using only the clinical pattern and three blood tests: total bilirubin, direct (conjugated) bilirubin, and indirect (unconjugated) bilirubin.
The complete pathway from haem catabolism to biliary excretion has four anatomically distinct steps, each of which can be disrupted:
Step 1 — Haem catabolism (pre-hepatic): In the reticuloendothelial system (spleen, bone marrow), senescent red blood cells are broken down. The haem ring is opened by haem oxygenase to produce biliverdin, which is reduced by biliverdin reductase to unconjugated bilirubin. This unconjugated bilirubin is water-insoluble, bound to albumin, and transported to the liver. When haem catabolism is accelerated — as in haemolytic anaemia (sickle cell disease, thalassaemia, G6PD deficiency, autoimmune haemolysis), ineffective erythropoiesis, or large haematoma resorption — the liver's conjugation capacity is exceeded, and unconjugated (indirect) bilirubin accumulates in plasma. This is pre-hepatic jaundice: unconjugated bilirubin dominant, urine bilirubin absent (because unconjugated bilirubin is not water-soluble and cannot be excreted by the kidney), urobilinogen in urine increased (because more conjugated bilirubin reaches the gut).
Step 2 — Hepatic uptake and conjugation (hepatic): After dissociating from albumin at the hepatocyte sinusoidal surface, unconjugated bilirubin is transported into the hepatocyte by OATP1B1/1B3 transporters and conjugated with glucuronic acid by UDP-glucuronosyltransferase 1A1 (UGT1A1) to form bilirubin mono- and di-glucuronide — the direct (conjugated) form. Defects at this step produce unconjugated hyperbilirubinaemia. The classic example is Gilbert syndrome (mild UGT1A1 promoter polymorphism, benign unconjugated hyperbilirubinaemia that worsens with fasting or illness) and Crigler-Najjar syndrome (complete UGT1A1 absence — severe, neonatal kernicterus). In generalised hepatocellular disease (viral hepatitis, cirrhosis), all steps are impaired and a mixed picture results.
Step 3 — Canalicular excretion (intrahepatic): Conjugated bilirubin is excreted into the bile canaliculus via the MRP2 (ABCC2) transporter. This is the energy-dependent, rate-limiting step. Defective MRP2 causes Dubin-Johnson syndrome (benign conjugated hyperbilirubinaemia, characteristic dark liver pigment). In hepatocellular disease, canalicular excretion is impaired alongside all other hepatocyte functions, contributing to conjugated hyperbilirubinaemia.
Step 4 — Bile flow and intestinal excretion (post-hepatic/cholestatic): Conjugated bilirubin passes through the bile duct system into the duodenum, where gut bacteria reduce it to urobilinogen (some reabsorbed and excreted in urine) and then to stercobilin (excreted in faeces, giving them their brown colour). Obstruction of bile flow — whether within the liver (intrahepatic cholestasis: primary biliary cholangitis, drug-induced cholestasis, viral hepatitis-induced cholestasis) or in the extrahepatic duct system (choledocholithiasis, pancreatic head carcinoma, cholangiocarcinoma) — traps conjugated bilirubin and causes cholestatic/post-hepatic jaundice: direct bilirubin dominant, dark urine (conjugated bilirubin is water-soluble and excreted by the kidney → bilirubinuria), pale stools (no stercobilin reaches gut), elevated serum alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) (markers of bile duct injury/induction), pruritus (bile salt retention).
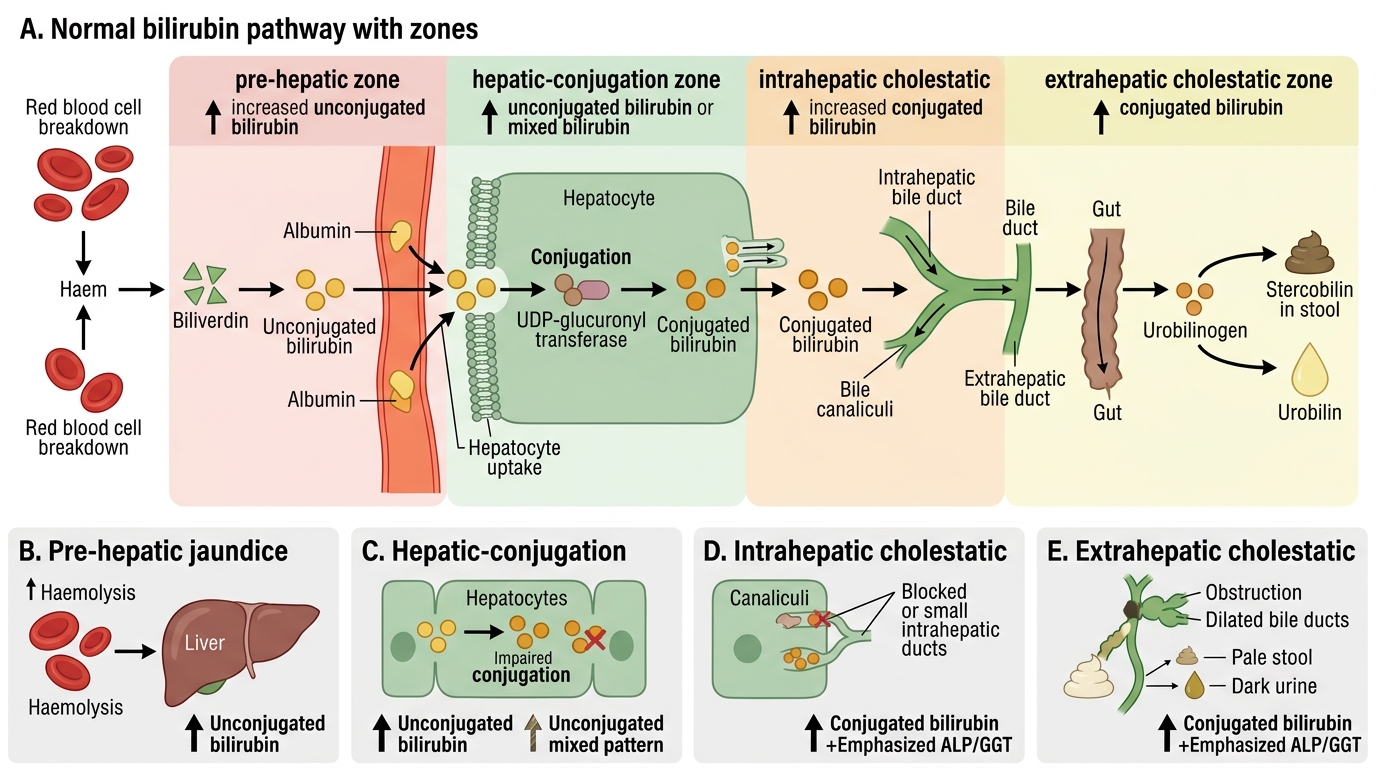
Bilirubin Metabolism and Patterns of Jaundice
Aetiology and Pathophysiology of Liver Disease
The liver can be damaged by a broad range of insults, each with a characteristic mechanism, cellular target, and histological pattern. Rather than memorising an undifferentiated list, organising the aetiologies by pathogenic mechanism provides a framework that makes the clinical picture predictable and the investigation findings interpretable. Every major liver disease has a fingerprint in the liver function tests — a combination of which enzymes are elevated, by how much, and in what ratio — that reflects the dominant form of hepatocyte injury. Hepatocellular injury (direct hepatocyte death or inflammation) elevates alanine aminotransferase (ALT) and aspartate aminotransferase (AST) disproportionately; cholestatic injury (obstruction of bile flow, intrahepatic or extrahepatic) elevates alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) disproportionately; and synthetic failure (reduced hepatocyte mass or function) manifests as low albumin, prolonged INR, and hypoglycaemia. Most clinical liver diseases show a mixed pattern, but the dominant pattern guides the initial differential diagnosis and the first investigation priority.
Provided image
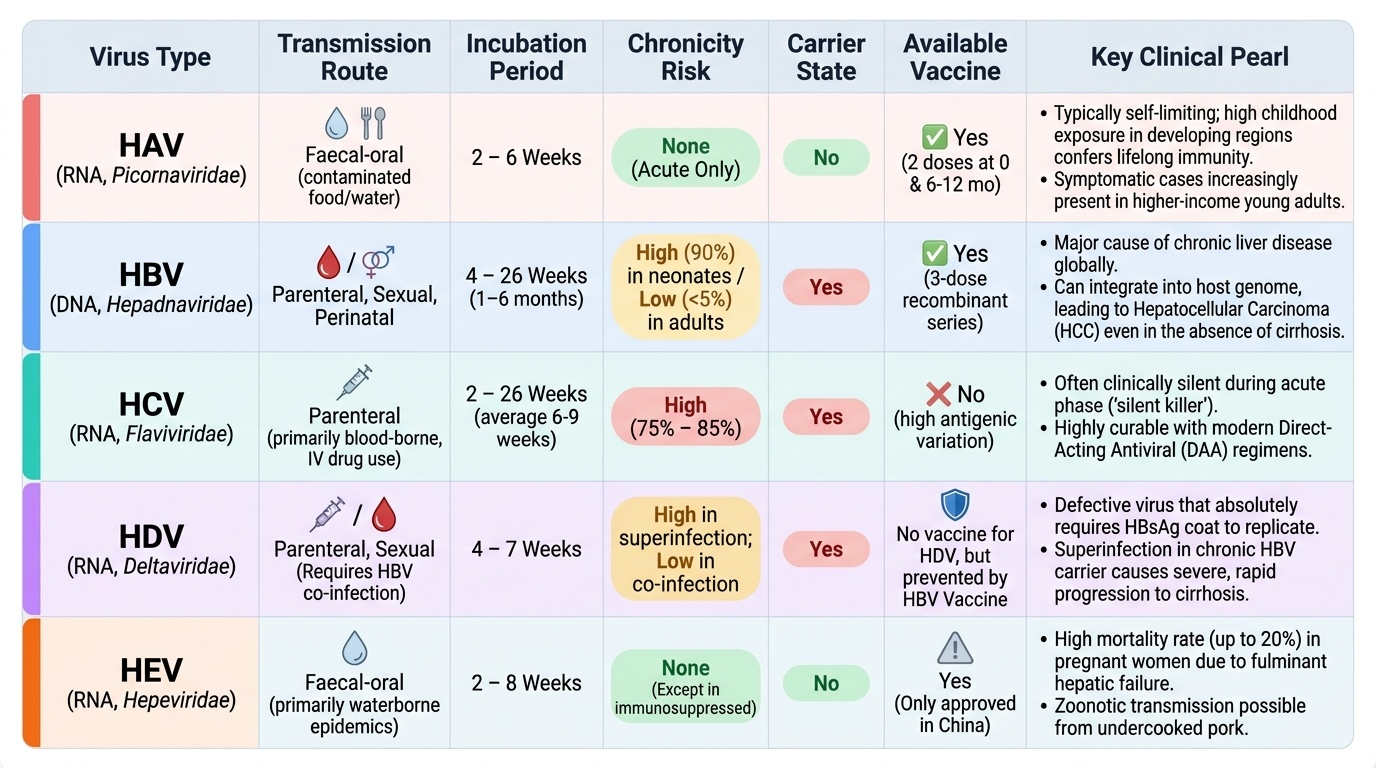
Viral hepatitis is the most important cause of acute and chronic liver disease globally and in India. The hepatitis viruses differ fundamentally in their mode of transmission, propensity for chronicity, and outcome:
- Hepatitis A virus (HAV): RNA virus (Picornaviridae), transmitted by the faecal-oral route (contaminated water and food). HAV causes acute hepatitis only — it does not cause chronic infection. The incubation period is 2–6 weeks. In India, seroprevalence from childhood exposure is high in low-income populations, conferring lifelong immunity, which explains why symptomatic infection increasingly presents in young adults from higher-income backgrounds who escaped natural exposure (as in Priya's case in the hook). HAV infection is typically self-limiting; fulminant hepatic failure occurs in <1% of cases but is more common in those with underlying liver disease. No chronic carrier state; no antiviral treatment — management is supportive. Prevention: hepatitis A vaccine (2 doses, 0 and 6–12 months).
- Hepatitis B virus (HBV): DNA virus (Hepadnaviridae), transmitted parenterally, sexually, and perinatally (mother-to-child transmission is the major route in endemic regions including India). Incubation: 1–6 months. The key distinction from HAV is chronicity: the probability of chronic infection depends on the age at acquisition — perinatal infection leads to chronic HBV in ~90% of cases (immune tolerance phase); adult-acquired infection leads to chronic HBV in only 5–10%. Chronic HBV infects approximately 40 million Indians. Hepatitis B immunisation (3-dose schedule at birth/1 month/6 months) is the most effective prevention; the birth dose within 24 hours is critical for blocking perinatal transmission.
- Hepatitis C virus (HCV): RNA virus (Flaviviridae), transmitted predominantly parenterally (intravenous drug use, needle-sharing, transfusion, haemodialysis, unsafe injections). Sexual and perinatal transmission occur but are less efficient than HBV. Acute HCV is often asymptomatic. Critically, chronicity develops in approximately 75–85% of acute HCV infections — far higher than HBV. No effective vaccine exists for HCV (this is a common trap: do not state or imply that an HCV vaccine exists).
- Hepatitis D virus (HDV): RNA virus, defective — requires HBsAg coat from HBV for assembly. Occurs only as co-infection with HBV (simultaneous) or superinfection (in a chronic HBV carrier). HDV superinfection dramatically accelerates cirrhosis progression.
- Hepatitis E virus (HEV): RNA virus, faecal-oral transmission. Typically self-limiting. The critical exception is pregnancy: HEV in the third trimester causes fulminant hepatic failure in 15–25% of cases — the highest case-fatality for any hepatitis virus in this population, highly relevant to Indian practice where waterborne outbreaks are documented.
Pathophysiology of viral hepatitis: The dominant mechanism of hepatocyte injury in HBV and HCV is immune-mediated cytotoxicity — not direct viral cytopathology. Virus-specific cytotoxic T lymphocytes (CD8+) recognise viral antigens presented on HLA class I molecules on infected hepatocytes and destroy them. This explains why immunosuppressed patients (HIV co-infection, transplant recipients) may sustain high viral replication loads with minimal liver inflammation yet progress to severe disease once immune reconstitution occurs. The histological hallmark of acute viral hepatitis is hepatocyte necrosis (lobular hepatitis) with lymphocytic infiltrate and the pathognomonic Councilman (acidophil) bodies — apoptotic hepatocytes with eosinophilic cytoplasm.
Alcoholic Liver Disease
Alcoholic liver disease (ALD) is the leading cause of cirrhosis in Indian tertiary hepatology centres and the archetype of toxic hepatocellular injury. The spectrum runs from steatosis (fatty liver) through alcoholic hepatitis to cirrhosis, and progression along this spectrum depends on quantity of alcohol consumed, pattern of drinking, genetic susceptibility, and co-existing metabolic or viral liver disease.
The central mechanism of alcohol-induced liver injury begins with hepatic ethanol metabolism. Ethanol is oxidised in the hepatocyte primarily by alcohol dehydrogenase (ADH) in the cytosol (and by MEOS — microsomal ethanol oxidising system, CYP2E1, at higher concentrations and with chronic use) to acetaldehyde, and then by aldehyde dehydrogenase (ALDH) to acetate. Both ADH and ALDH reactions reduce NAD+ to NADH, profoundly shifting the cellular NAD+/NADH ratio. This metabolic shift has several downstream consequences: impaired fatty acid oxidation (reduced beta-oxidation because beta-oxidation requires NAD+), increased fatty acid synthesis (NADH promotes lipogenesis), and impaired gluconeogenesis (gluconeogenic substrates like lactate and pyruvate shunted toward NADH-accepting reactions). The result is steatosis — triglyceride accumulation in hepatocytes, visible on histology as macrovesicular fat droplets. Steatosis is reversible with abstinence.
With continued alcohol use, acetaldehyde — the highly reactive intermediate — drives the next stage of injury. Acetaldehyde forms protein adducts that activate hepatic stellate cells (perisinusoidal cells of Ito), promotes oxidative stress via reactive oxygen species generated by CYP2E1, activates the NLRP3 inflammasome, and stimulates Kupffer cell (hepatic macrophage) production of TNF-alpha and IL-1beta through lipopolysaccharide (LPS) derived from increased intestinal permeability — the gut-liver axis plays a central role. This inflammatory milieu causes alcoholic hepatitis: histologically characterised by hepatocyte ballooning degeneration, Mallory-Denk bodies (aggregates of ubiquitinated cytokeratin filaments), neutrophilic infiltration (distinguishing alcoholic from viral hepatitis), and pericellular fibrosis around centrilobular hepatocytes (zone 3 — centrilobular necrosis is characteristic of ALD, whereas viral hepatitis preferentially damages the periportal zone).
Alcoholic hepatitis is a distinct clinical syndrome: acute onset of jaundice, fever, hepatomegaly, and elevated serum AST:ALT ratio >2:1 (because alcohol induces mitochondrial damage preferentially reducing ALT; also alcohol depletes pyridoxal-5'-phosphate, a cofactor needed more for ALT activity). Discriminant function (DF) = 4.6 × (PT patient − PT control) + bilirubin (mg/dL); a score >32 indicates severe alcoholic hepatitis with >35% 1-month mortality — this threshold indicates a possible role for corticosteroids (prednisolone 40 mg/day × 28 days) provided infection has been excluded. The MELD score (see cirrhosis section) correlates with 90-day mortality in ALD.
Progressive fibrosis leads to cirrhosis: replacement of normal architecture by regenerative nodules surrounded by fibrous septa — an irreversible structural change. The trigger for fibrosis is persistent hepatic stellate cell activation by alcohol metabolites, oxidative stress, and pro-fibrotic cytokines (TGF-beta is the master pro-fibrotic cytokine).
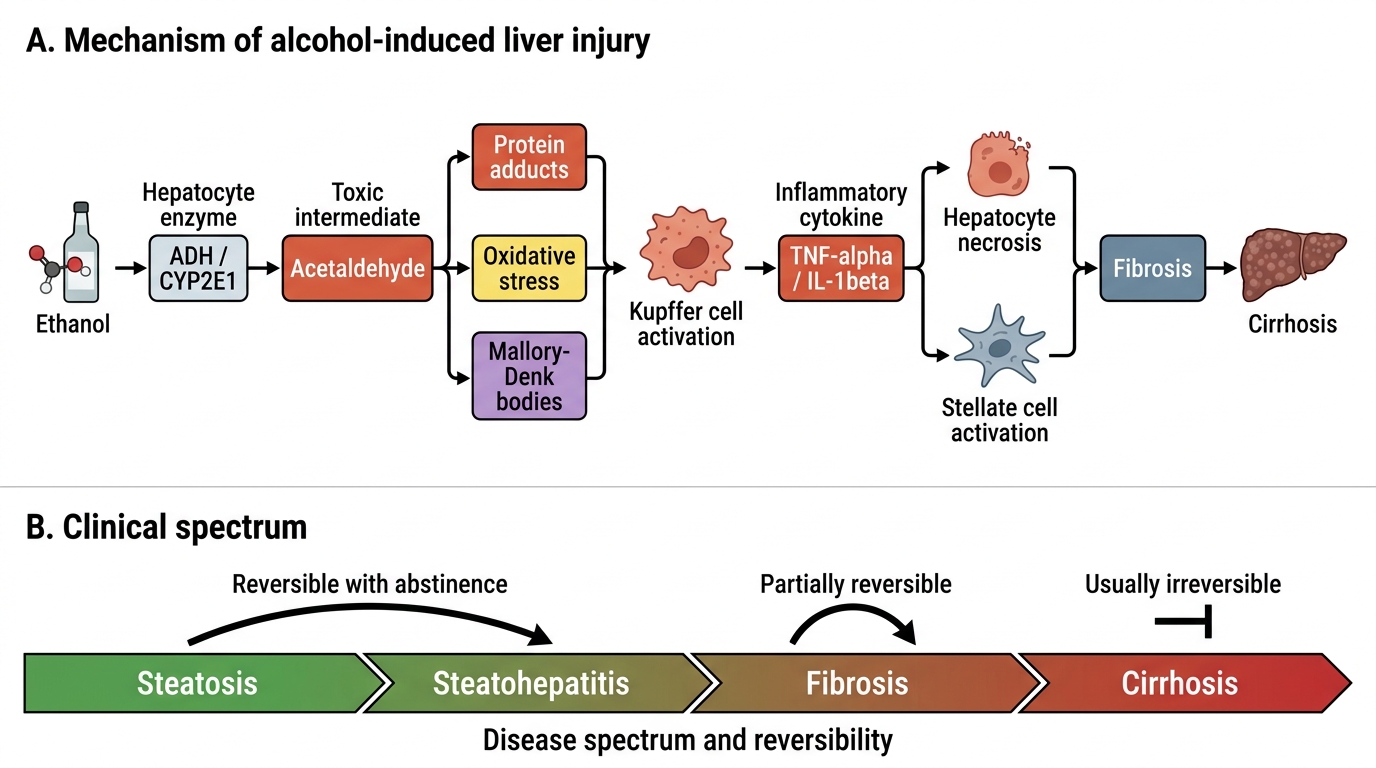
Alcohol-Induced Liver Injury Mechanism