Page 2 of 19
IM5.1-7 | Liver Disease Foundations — SDL Guide (Part 2)
Cirrhosis and Portal Hypertension
Cirrhosis represents the final common pathway of virtually all chronic liver diseases. It is defined histologically as diffuse hepatic fibrosis with formation of regenerative nodules that disrupt the normal lobular architecture. Once established, cirrhosis produces two fundamental consequences — hepatocellular failure (loss of metabolic and synthetic function) and portal hypertension (elevated pressure in the portal venous system) — and it is the complications arising from these two mechanisms that cause morbidity and mortality.
The functional staging of cirrhosis determines prognosis and guides management decisions. Two validated scoring systems are used:
Child-Pugh score was developed to stratify operative risk in cirrhotic patients undergoing portosystemic shunt surgery. It incorporates exactly five parameters: serum bilirubin (mg/dL), serum albumin (g/dL), INR/prothrombin time, ascites (none/mild/moderate–severe), and hepatic encephalopathy (none/grade I–II/grade III–IV). Each parameter is scored 1–3, for a total of 5–15 points: Class A (5–6) = well-compensated, 1-year survival ~100%; Class B (7–9) = significant dysfunction, 1-year survival ~80%; Class C (10–15) = decompensated, 1-year survival ~45%. Note that Child-Pugh includes clinical parameters (ascites and encephalopathy) alongside laboratory values, making it somewhat subjective.
MELD score (Model for End-Stage Liver Disease) was derived and validated to predict 90-day mortality in cirrhotic patients, replacing Child-Pugh as the standard for liver transplant allocation. MELD is calculated from three objective laboratory values: serum bilirubin (mg/dL), serum creatinine (mg/dL), and INR. The original MELD formula is: MELD = 9.57 × ln(creatinine) + 3.78 × ln(bilirubin) + 11.20 × ln(INR) + 6.43. MELD-Na adds serum sodium to improve prediction in patients with hyponatraemia. A MELD score ≥15 generally indicates that liver transplant benefit outweighs surgical risk. The critical distinction: Child-Pugh contains albumin and clinical ascites/encephalopathy; MELD contains creatinine but not albumin — these parameters must never be confused between the two systems.
Portal hypertension arises when sinusoidal resistance increases (due to fibrosis and regenerative nodule compression of hepatic venules) and splanchnic blood flow increases (due to portosystemic shunting and splanchnic vasodilatation mediated by nitric oxide and glucagon). Portal pressure is typically expressed as the hepatic venous pressure gradient (HVPG) — normal <5 mmHg; clinically significant portal hypertension ≥10 mmHg (point at which varices form); severe ≥12 mmHg (threshold for variceal haemorrhage and ascites).
The major complications of cirrhosis — each with a distinct pathophysiological mechanism — are:
- Variceal haemorrhage: Portosystemic collaterals develop as portal pressure rises, particularly at the gastro-oesophageal junction (oesophageal varices — the most dangerous), the gastric fundus, rectum (haemorrhoids), paraumbilical veins (caput medusae), and retroperitoneum. Variceal haemorrhage is precipitated when portal pressure exceeds the transmural pressure the variceal wall can sustain (threshold HVPG ~12 mmHg). Mortality from acute variceal bleed is 15–20% at 6 weeks.
- Ascites: Portal hypertension raises hydrostatic pressure in the splanchnic capillaries, promoting transudation of fluid into the peritoneal cavity. Concurrently, hypoalbuminaemia (due to failed hepatic synthesis) reduces plasma oncotic pressure. Activation of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system in response to splanchnic vasodilatation causes avid renal sodium and water retention, perpetuating ascites. The SAAG (serum-ascites albumin gradient) = serum albumin − ascites albumin; SAAG ≥ 1.1 g/dL indicates portal hypertension with high sensitivity (>97%); SAAG < 1.1 g/dL indicates a non-portal aetiology (TB peritonitis, malignant ascites, nephrotic syndrome, pancreatitis).
- Spontaneous bacterial peritonitis (SBP): Bacterial translocation from the gut (particularly gram-negative organisms, predominantly Escherichia coli, Klebsiella pneumoniae, Streptococcus pneumoniae) through a compromised intestinal mucosal barrier infects ascitic fluid. Diagnostic criterion: ascitic fluid polymorphonuclear (PMN) cell count ≥ 250 cells/mm³ — this threshold triggers empirical treatment even before culture results are available, because culture sensitivity is only 40–50%. SBP is a marker of severely decompensated disease with 1-year mortality ~50%.
- Hepatic encephalopathy (HE): Failure of the cirrhotic liver to clear gut-derived nitrogenous waste products — principally ammonia (produced by intestinal bacterial urease and glutaminase acting on urea and amino acids) — leads to astrocyte swelling and cerebral dysfunction. The West-Haven grading system classifies HE severity: Grade I (subtle personality changes, impaired concentration), Grade II (obvious asterixis, confusion, disorientation), Grade III (somnolence, gross confusion, incoherent speech — arousable), Grade IV (coma — not arousable). Precipitating factors: GI bleed (protein load), infection, hyponatraemia, constipation, diuretic excess, sedatives, hypoglycaemia. Asterixis (hepatic flap) — the inability to maintain a sustained posture, demonstrated by asking the patient to extend arms with hands dorsiflexed — is the cardinal sign of HE grade II.
- Hepatorenal syndrome (HRS): Functional renal failure in cirrhotic patients with ascites, caused by profound splanchnic vasodilatation (mediated by vasodilators including nitric oxide and prostacyclin) that activates renal vasoconstriction via the RAAS and sympathetic system, reducing glomerular filtration rate in the absence of intrinsic renal disease. Type 1 (HRS-AKI): rapid deterioration, creatinine doubling to >2.5 mg/dL within 2 weeks, triggered by SBP or other precipitants — median survival without liver transplant is weeks. Type 2 (HRS-CKD): more gradual, associated with refractory ascites. Diagnosis requires exclusion of other causes of AKI (pre-renal, obstructive, parenchymal) and lack of response to intravenous albumin plus 48-hour diuretic withdrawal. Treatment: terlipressin + albumin is the first-line pharmacological therapy (terlipressin — a vasopressin analogue — causes splanchnic vasoconstriction and redirects blood flow to the kidney).
- Hepatocellular carcinoma (HCC): Cirrhosis is the most important risk factor for HCC regardless of the underlying aetiology; HBV-related HCC can arise even in the absence of established cirrhosis (an important exception). HCC complicates cirrhosis at a rate of 2–4% per year. Screening: 6-monthly liver ultrasound ± AFP in all cirrhotic patients and in high-risk HBV carriers. Alpha-fetoprotein (AFP) >400 ng/mL in a cirrhotic patient is strongly diagnostic. Staging by the Barcelona Clinic Liver Cancer (BCLC) system integrates tumour stage, liver function (Child-Pugh), and performance status to guide treatment (surgical resection, ablation, transarterial chemoembolisation — TACE, sorafenib/lenvatinib, liver transplantation within Milan criteria).
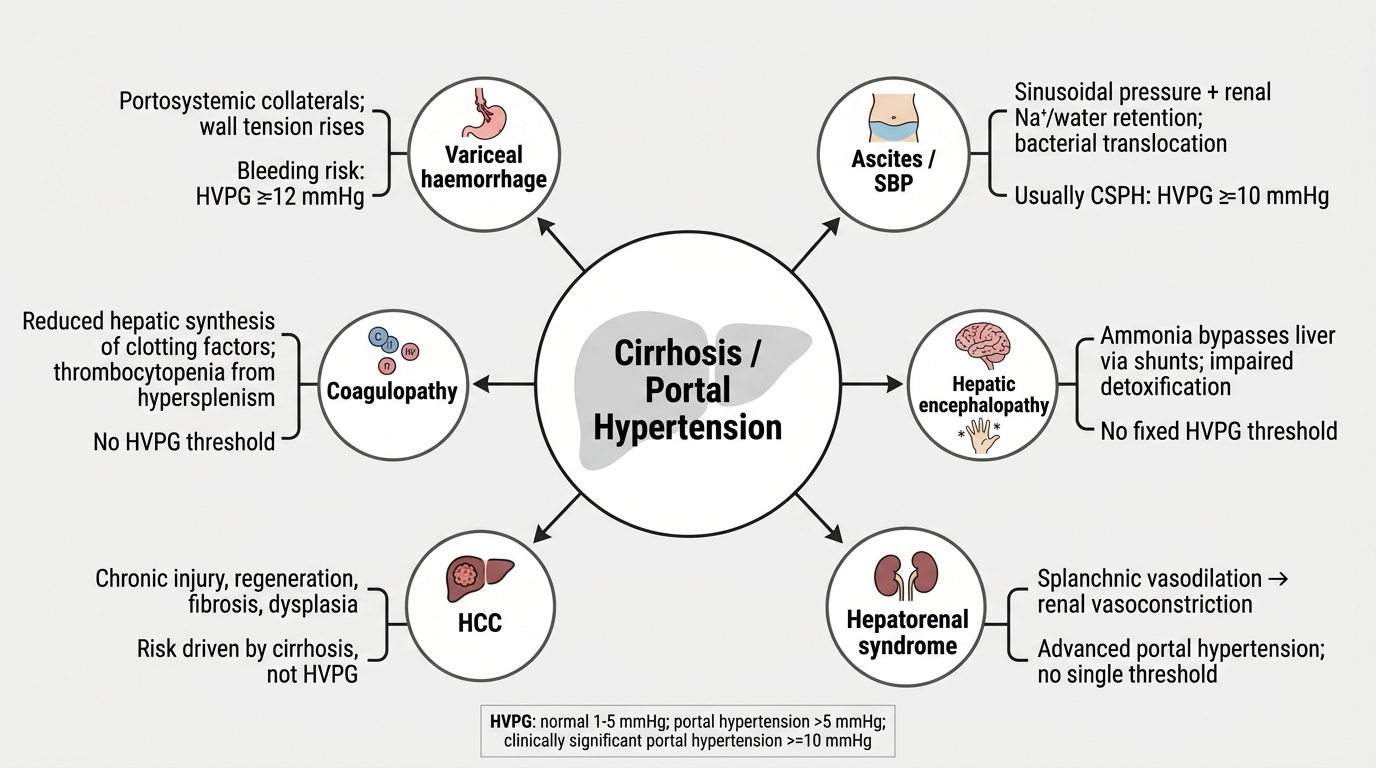
Complications of Cirrhosis and Portal Hypertension
SELF-CHECK
A 45-year-old man with known cirrhosis presents with confusion and asterixis. Serum ammonia is elevated. His Child-Pugh score is being calculated. Which of the following parameters is included in Child-Pugh ONLY, and NOT in the MELD score?
A. Serum bilirubin
B. INR
C. Serum albumin
D. Serum creatinine
Reveal Answer
Answer: C. Serum albumin
The Child-Pugh score incorporates five parameters: bilirubin, albumin, INR/PT, ascites, and hepatic encephalopathy. The MELD score uses only three objective values: bilirubin, creatinine, and INR. Serum albumin is ONLY in Child-Pugh; serum creatinine is ONLY in MELD. Both scores include bilirubin and INR. A common error is putting creatinine into Child-Pugh or albumin into MELD — this question tests that distinction directly.
Drug-Induced Liver Injury
Drug-induced liver injury (DILI) is a heterogeneous group of hepatic reactions caused by medications, herbal preparations, dietary supplements, and occupational toxins. It is the most common cause of acute liver failure in the United States and a frequent cause in India (where traditional herbal medicines — Ayurvedic preparations — are a significant additional source). DILI accounts for approximately 5–10% of all hospital admissions for jaundice and is frequently underdiagnosed because its presentation can mimic virtually any other liver disease.
DILI is broadly classified into two mechanistic categories based on predictability:
Intrinsic (dose-dependent, predictable) DILI occurs in any individual exposed to a sufficient dose of the hepatotoxic agent. The prototype is paracetamol (acetaminophen) hepatotoxicity — the most common cause of acute liver failure from DILI worldwide. At therapeutic doses (≤4 g/day in healthy adults, lower in alcohol users or malnourished patients), >95% of paracetamol is glucuronidated or sulphated and excreted harmlessly. A small fraction is metabolised by CYP2E1 (predominantly) and CYP3A4 to the highly reactive electrophile N-acetyl-para-benzoquinoneimine (NAPQI). NAPQI is normally detoxified by conjugation with glutathione in the hepatocyte. In overdose, glucuronidation and sulphation pathways are saturated, NAPQI production overwhelms glutathione stores, and NAPQI binds to hepatocyte mitochondrial proteins, causing oxidative stress, mitochondrial failure, and centrilobular (zone 3) necrosis. Risk factors for paracetamol hepatotoxicity include: chronic alcoholism (CYP2E1 induction + glutathione depletion), malnutrition, concurrent inducers of CYP2E1, and isoniazid therapy. Treatment: N-acetylcysteine (NAC), which replenishes glutathione precursors. The Rumack-Matthew nomogram (serum paracetamol level plotted against time since ingestion) is used to stratify risk and guide NAC administration after acute overdose.
Idiosyncratic DILI is unpredictable, not dose-dependent, and occurs in susceptible individuals based on genetic factors (HLA haplotypes, polymorphisms in metabolising enzymes and transporters), immune dysregulation, or individual metabolic idiosyncrasy. Latency from drug exposure to injury is variable (days to months). The main mechanisms are (a) immune-mediated: reactive drug metabolites bind to hepatocyte proteins, creating neo-antigens that trigger an adaptive immune response (cytotoxic T cells, antibodies) — this explains the occasional eosinophilia, skin rash, and fever (drug hypersensitivity hepatitis); and (b) metabolic idiosyncrasy: individual variation in drug metabolism leads to accumulation of hepatotoxic intermediates.
Important drugs causing DILI that every clinician must know:
| Drug | Pattern | Mechanism | Notes |
|---|---|---|---|
| Paracetamol | Hepatocellular, zone 3 necrosis | Intrinsic (NAPQI) | Threshold dose, treat with NAC |
| Isoniazid (INH) | Hepatocellular | Idiosyncratic metabolic | Monitor LFTs; incidence higher with rifampicin combination; age-related |
| Rifampicin | Cholestatic/mixed | Transporter inhibition | Usually with INH; causes unconj. hyperbilirub. by UGT competition |
| Methotrexate | Fibrosis/cirrhosis | Cumulative dose | Folate depletion; liver biopsy at cumulative dose thresholds |
| Amiodarone | Steatohepatitis (NASH-like) | Mitochondrial | Phospholipidosis; can mimic alcoholic hepatitis |
| Statins | Hepatocellular (rare, mild) | Idiosyncratic | Transaminase elevation; rarely severe; safe to use with mild elevation |
| Traditional herbal medicines | Variable | Pyrrolizidine alkaloids, heavy metals | Kava, comfrey, senecio cause sinusoidal obstruction syndrome; Ayurvedic preparations |
The RUCAM (Roussel Uclaf Causality Assessment Method) score is the internationally validated tool for assessing causality in suspected DILI — it assigns points for temporal association, risk factors, exclusion of other causes, response to dechallenge (drug withdrawal) and rechallenge, and known hepatotoxic potential of the drug.
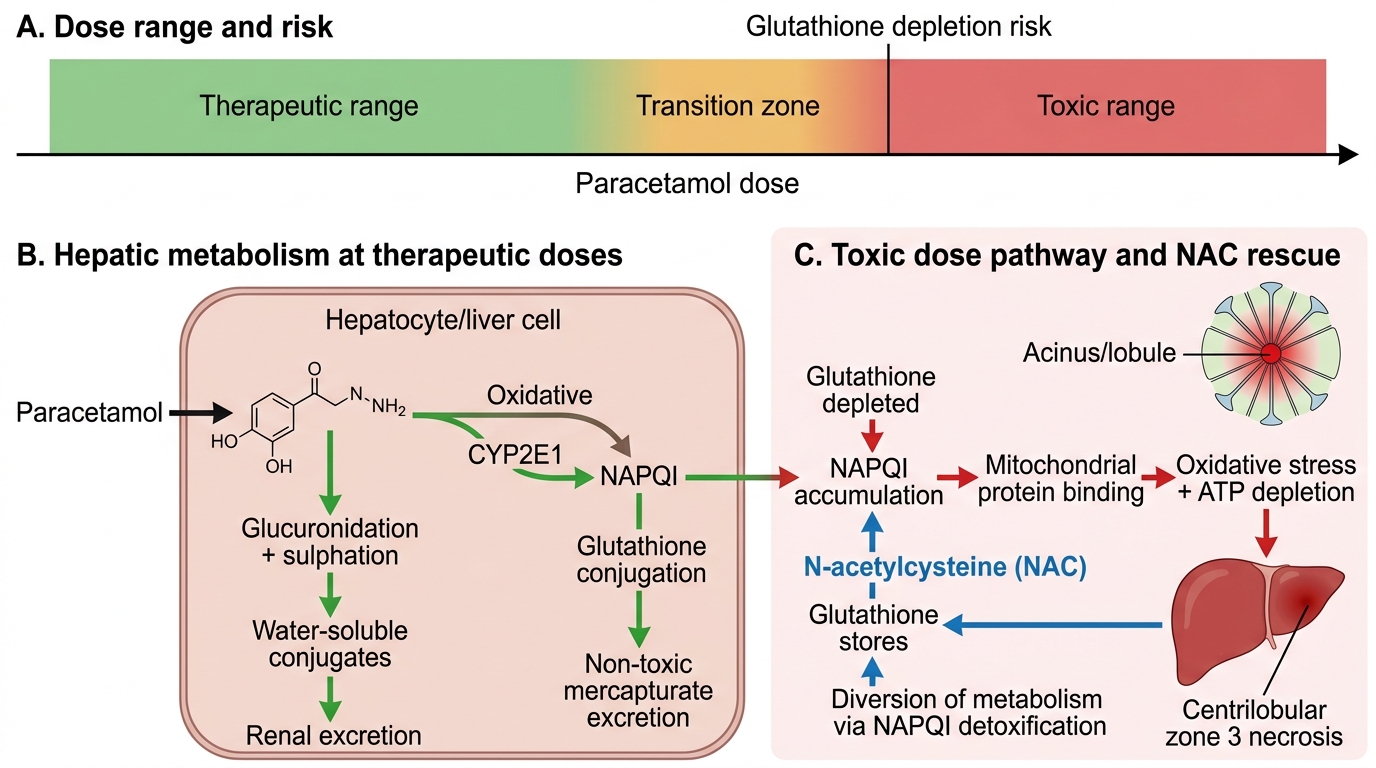
Mechanism of Paracetamol Hepatotoxicity
Cholelithiasis and Cholecystitis
Cholelithiasis (gallstone disease) and its principal complication, cholecystitis, are among the most common surgical conditions in adults, but they present to internists with jaundice, biliary colic, and acute abdominal pain and have important medical management considerations. Understanding their pathophysiology and complications is therefore an essential competency for the general physician.
Gallstones form when bile composition is altered such that normally soluble components precipitate. The two principal stone types have distinct compositions and pathogeneses:
Cholesterol stones (80% of gallstones in Western populations; also predominant in India among urban, higher-income groups) form when bile is supersaturated with cholesterol relative to bile salts and phospholipids. The critical determinant is the cholesterol saturation index (CSI): when CSI >1, bile is lithogenic. Three conditions promote cholesterol supersaturation: (1) excess hepatic cholesterol secretion into bile (obesity, diabetes, dyslipidaemia, pregnancy — elevated oestrogen increases hepatic cholesterol synthesis; oral contraceptives); (2) reduced bile salt pool (ileal disease or resection — bile salts are actively reabsorbed in the terminal ileum, so ileal pathology reduces the recycled pool; Crohn disease); (3) gallbladder hypomotility (pregnancy, prolonged fasting, total parenteral nutrition, somatostatin therapy — inadequate gallbladder contraction allows bile to stagnate and cholesterol to nucleate). Additional factor: mucin glycoproteins secreted by the gallbladder mucosa act as nucleating agents accelerating crystal formation.
Pigment stones are either black (bilirubin polymer, form in the gallbladder in haemolytic states — sickle cell disease, hereditary spherocytosis, thalassaemia — when excess unconjugated bilirubin is secreted into bile; also cirrhosis) or brown (calcium bilirubinate + fatty acid salts, form in bile ducts and are associated with bacterial infection of bile — E. coli bacterial beta-glucuronidase deconjugates bilirubin glucuronide → free unconjugated bilirubin precipitates; more common in Southeast Asian populations and in the setting of biliary stasis from strictures or parasites).
Clinical spectrum of cholelithiasis:
- Asymptomatic gallstones: The majority (~80%) of gallstones never cause symptoms. Risk of developing symptoms is approximately 2–3% per year; prophylactic cholecystectomy is not routinely recommended for asymptomatic stones.
- Biliary colic: Transient obstruction of the cystic duct by a gallstone produces episodic, severe right upper quadrant (RUQ) or epigastric pain, often radiating to the right scapula, typically triggered by fatty meals (which stimulate cholecystokinin release, causing gallbladder contraction). The pain builds over 15–30 minutes, is constant (despite the name 'colic'), lasts 1–5 hours, and resolves as the stone falls back into the gallbladder. Liver function tests are normal during an uncomplicated episode; ultrasound is the investigation of choice (sensitivity >95% for gallstones).
- Acute cholecystitis: Persistent cystic duct obstruction leads to gallbladder distension, mucosal ischaemia, secondary bacterial infection, and inflammation. Murphy's sign (inspiratory arrest on RUQ palpation during inspiration) is the classic clinical sign. Laboratory features: leukocytosis, mildly elevated bilirubin (<3 mg/dL; higher levels suggest choledocholithiasis or Mirizzi syndrome), elevated ALP and GGT. Ultrasound shows gallbladder wall thickening (>4 mm), pericholecystic fluid, and a positive sonographic Murphy's sign. Management: fasting, analgesia, IV antibiotics (to cover gram-negative enteric organisms and anaerobes), and cholecystectomy (laparoscopic — the gold standard, performed within 72 hours of admission in uncomplicated acute cholecystitis).
- Choledocholithiasis (common bile duct stones): Stones passing from the gallbladder into the common bile duct cause obstructive jaundice (progressively elevated direct bilirubin, elevated ALP, pale stools, dark urine, pruritus), and if the duct becomes infected, ascending cholangitis — Charcot's triad of fever, jaundice, and RUQ pain (and in severe cases, Reynolds' pentad adding shock and altered consciousness). ERCP with sphincterotomy and stone extraction is the definitive treatment for choledocholithiasis.
- Gallstone pancreatitis: Stones passing through the ampulla of Vater trigger pancreatitis by temporarily obstructing pancreatic duct drainage — the commonest cause of acute pancreatitis in women and the second commonest overall (after alcohol in men).
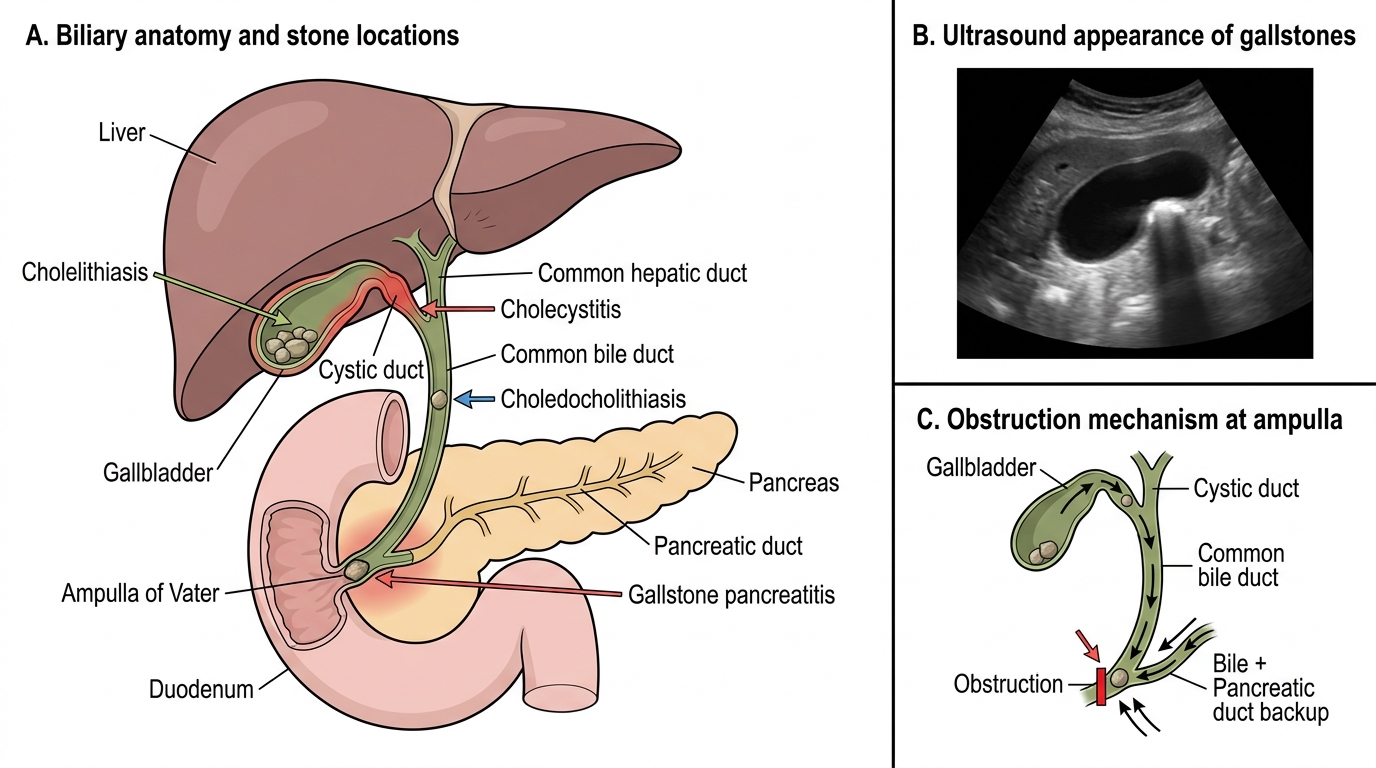
Biliary Stone Disease: Anatomy and Ultrasound Features
SELF-CHECK
A 32-year-old woman presents with acute upper abdominal pain, fever, and jaundice. She has Charcot's triad. Ultrasound shows gallstones and a dilated common bile duct. Which of the following describes the SAAG result you would expect if she also develops ascites from this condition?
A. SAAG ≥ 1.1 g/dL — portal hypertension aetiology
B. SAAG < 1.1 g/dL — non-portal aetiology (biliary peritonitis)
C. SAAG will be equal to serum albumin in biliary ascites
D. SAAG is not applicable to ascites from cholecystitis
Reveal Answer
Answer: B. SAAG < 1.1 g/dL — non-portal aetiology (biliary peritonitis)
SAAG (serum albumin − ascites albumin) ≥ 1.1 g/dL indicates portal hypertension as the cause of ascites (cirrhosis, right heart failure, Budd-Chiari). SAAG < 1.1 g/dL indicates a non-portal aetiology. Bile peritonitis from biliary perforation, or ascites from peritoneal spread of biliary infection, would produce a protein-rich exudate with SAAG < 1.1 g/dL — because the pathological process (biliary leak, inflammation) increases ascitic protein content, bringing it close to serum albumin and reducing the gradient. TB peritonitis, malignant ascites, and pancreatitis all have SAAG < 1.1 g/dL for the same reason.