Page 3 of 19
IM5.1-7 | Liver Disease Foundations — SDL Guide (Part 3)
Diagnostic Approach to Liver Disease
The diagnostic investigation of liver disease follows a structured, stepwise approach that moves from biochemical pattern recognition to specific aetiological testing. Rather than ordering a broad panel of tests without a working hypothesis, the skilled clinician uses the liver function test (LFT) pattern — hepatocellular versus cholestatic — to generate a short differential diagnosis before committing to imaging or serological panels. This stepwise approach is both more efficient and more cost-effective in resource-limited settings, and it is the approach expected at the NMC KH/SH level of competency. The LFT pattern reflects the dominant mechanism of injury: transaminase (ALT/AST) elevation indicates hepatocyte death or necrosis; alkaline phosphatase (ALP) and GGT elevation indicates damage to or obstruction of the bile canalicular system; and synthetic parameter failure (low albumin, prolonged INR) indicates loss of functional hepatocyte mass. This three-axis interpretation — hepatocellular injury, cholestasis, and synthetic function — is the clinical anchor that every subsequent investigation refines. The history and physical examination add the epidemiological and clinical context (alcohol use, parenteral exposures, travel, family history, drug history including herbal medicines, signs of chronic liver disease) that converts the pattern into a differential diagnosis.
Step 1 — Identify the biochemical pattern:
- Hepatocellular pattern: predominantly elevated ALT and AST (>3× upper limit of normal) with relatively preserved ALP. Causes: viral hepatitis (acute and chronic), alcoholic hepatitis (AST:ALT >2:1), NAFLD/NASH, autoimmune hepatitis, drug-induced hepatocellular injury (paracetamol, INH), Wilson disease, ischaemic hepatitis.
- Cholestatic pattern: predominantly elevated ALP and GGT with relatively preserved transaminases. Causes: extrahepatic obstruction (choledocholithiasis, pancreatic head tumour, cholangiocarcinoma, stricture) or intrahepatic cholestasis (primary biliary cholangitis, primary sclerosing cholangitis, drug-induced cholestasis, intrahepatic cholestasis of pregnancy).
- Mixed pattern: both transaminases and ALP elevated — common in acute viral hepatitis with cholestasis, some drug reactions, and sepsis-associated cholestasis.
- Synthetic failure: low albumin, prolonged INR/PT, hypoglycaemia — indicates significant hepatocyte mass loss, seen in acute liver failure, cirrhosis, or prolonged severe hepatitis.
Step 2 — Hepatitis serology (directed by history): For viral hepatitis, the serology sequence is:
- HBsAg: surface antigen — present in acute and chronic HBV infection; cleared in resolved infection.
- Anti-HBc IgM: IgM antibody to core antigen — marker of acute HBV infection (most sensitive marker in the window period when HBsAg may be declining).
- HBeAg: e-antigen — marker of high replication and infectivity; its clearance and anti-HBe seroconversion signals immune control.
- HBV DNA: quantitative PCR — gold standard for replication status; used to monitor antiviral therapy.
- Anti-HCV (ELISA): screening test for HCV exposure; if positive, confirm with HCV RNA (PCR) to distinguish active infection from past cleared infection (anti-HCV persists lifelong after resolved HCV, but HCV RNA is only detectable in active infection).
- Anti-HAV IgM: confirms acute hepatitis A.
- Anti-HEV IgM: for HEV in appropriate epidemiological context (waterborne outbreak, pregnancy).
Step 3 — Imaging:
- Ultrasound abdomen is the first-line imaging for all liver disease: identifies hepatomegaly, echogenicity (fatty liver = bright/echogenic; cirrhosis = coarsened, irregular, shrunken), portal hypertension features (splenomegaly, dilated portal/splenic veins, ascites), focal lesions (HCC, metastasis), and biliary system (gallstones, duct dilatation).
- MRCP (MR Cholangiopancreatography): non-invasive, gold-standard imaging for biliary tree anatomy — preferred when extrahepatic obstruction is suspected but ERCP is not immediately needed; excellent for choledocholithiasis, strictures, PSC (beaded bile ducts), and hilar cholangiocarcinoma.
- ERCP (Endoscopic Retrograde Cholangiopancreatography): invasive, therapeutic as well as diagnostic — indicated when intervention is planned (stone extraction, stenting for malignant obstruction, brush cytology for biliary stricture). Should not be used as a purely diagnostic tool when MRCP is available.
- CT abdomen (triphasic/contrast-enhanced): for focal liver lesions (HCC characterisation — arterial enhancement + portal washout = hallmark of HCC), staging malignancy, and when ultrasound is technically inadequate.
- FibroScan (transient elastography): non-invasive assessment of hepatic fibrosis severity by measuring liver stiffness; reduces the need for liver biopsy in staging chronic viral hepatitis and NAFLD.
Step 4 — Liver biopsy: reserved for situations where non-invasive tests are insufficient — grading/staging of chronic hepatitis, diagnosing autoimmune hepatitis, confirming NAFLD/NASH, evaluating unexplained hepatomegaly, and assessing cumulative drug toxicity (methotrexate). Contraindicated in uncorrectable coagulopathy, uncooperative patient, or suspected haemangioma.
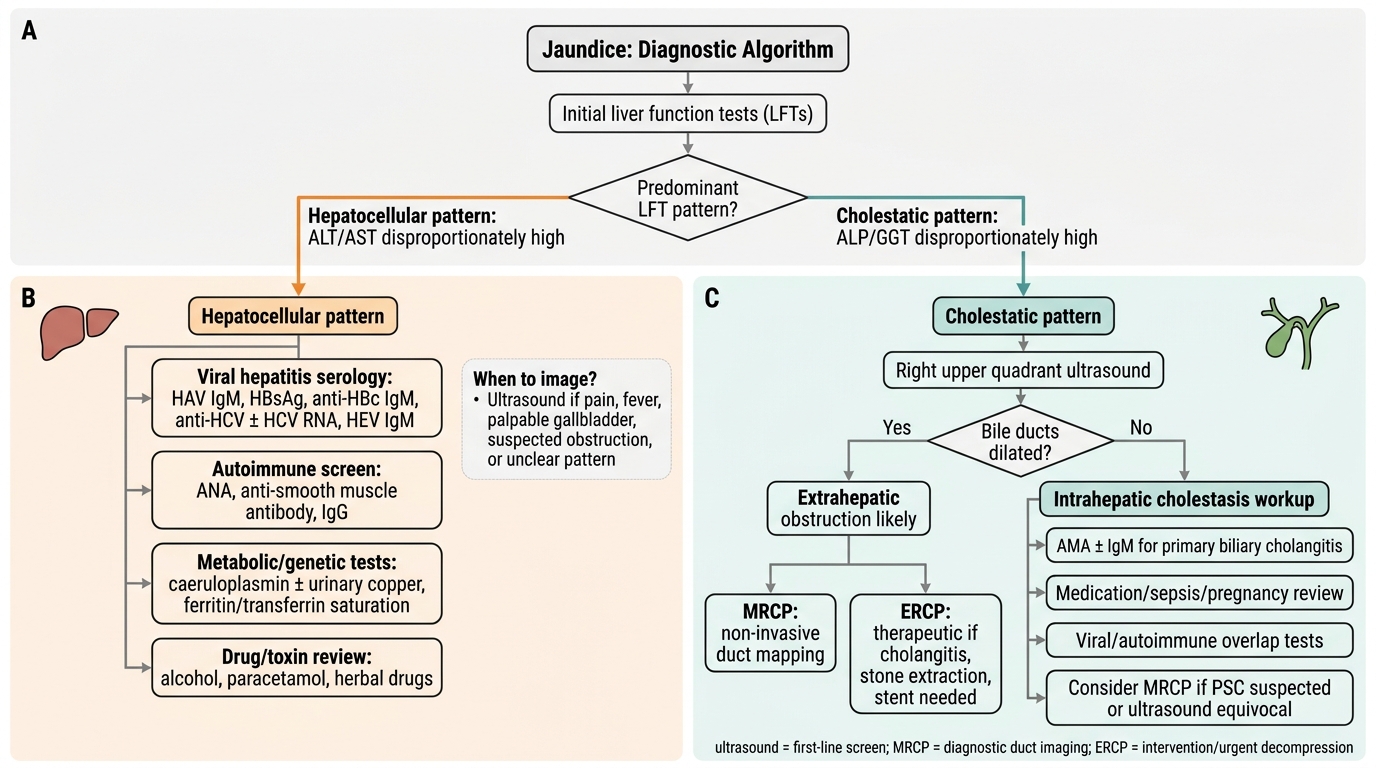
Diagnostic Algorithm for Jaundice
Principles of Management of Liver Disease
Management in hepatology is governed by three overarching principles that apply regardless of the specific aetiology: first, treat the underlying cause wherever a disease-modifying intervention exists; second, prevent and manage complications of liver disease — particularly those that are life-threatening and time-sensitive (variceal haemorrhage, SBP, HRS, acute liver failure); and third, counsel and plan for the long term — including cirrhosis surveillance, HCC screening, vaccination, and discussion of liver transplantation when appropriate. This module establishes the framework; detailed management of each condition is covered in subsequent SDL modules.
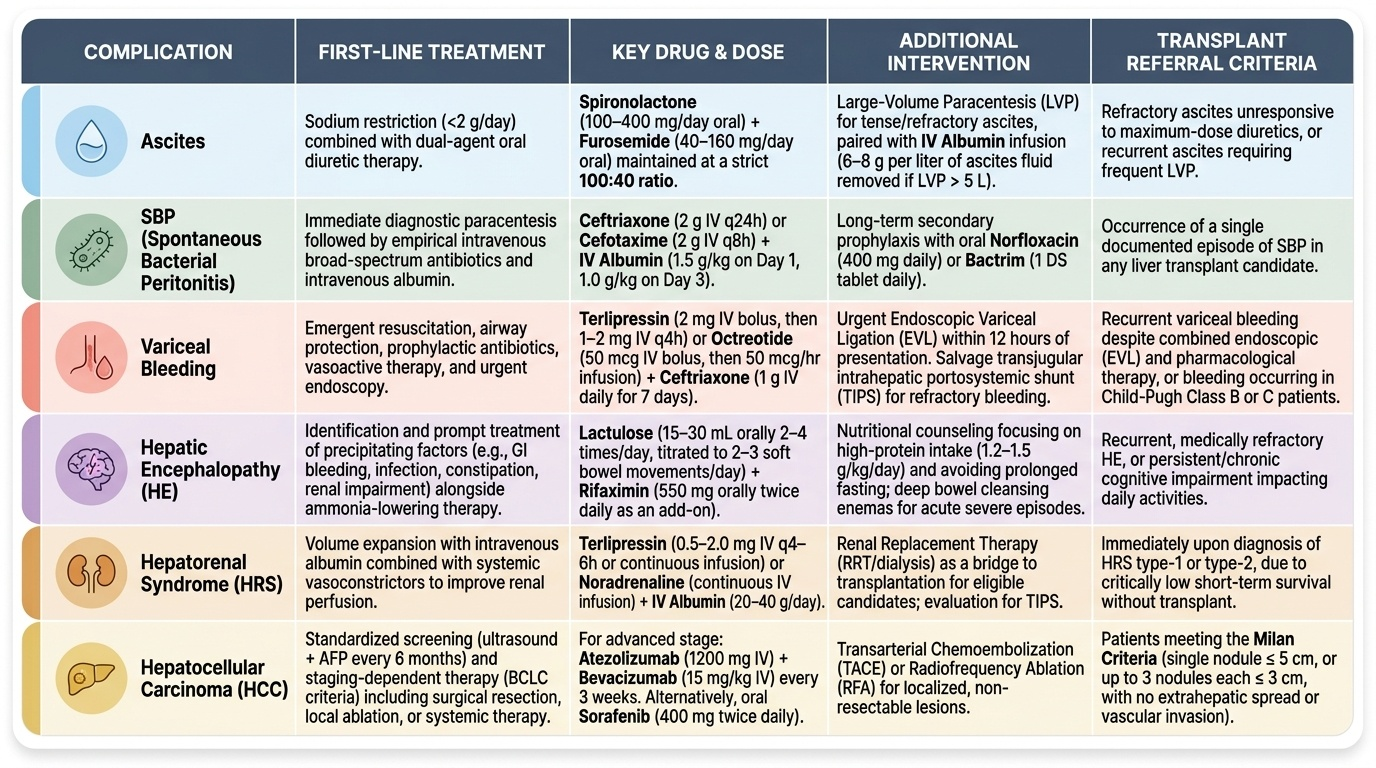
Provided image
Acute viral hepatitis: For HAV and HEV, management is entirely supportive — bed rest, oral hydration, avoidance of alcohol and hepatotoxic drugs, and nutritional support. No antiviral therapy is indicated. Hospital admission is required for severe hepatitis (INR >1.5, bilirubin >10 mg/dL, altered consciousness) or inability to maintain oral intake. For acute HBV in adults, >95% resolve spontaneously and antiviral treatment is not routinely indicated unless the patient is immunocompromised, develops acute liver failure, or requires immunosuppressive treatment for another condition. For acute HCV, guidelines increasingly recommend early direct-acting antiviral (DAA) therapy to prevent chronicity.
Chronic HBV: Antiviral therapy is indicated when HBV DNA is elevated and there is evidence of significant liver disease (raised ALT, significant fibrosis). First-line agents are tenofovir disoproxil fumarate (TDF) or entecavir — both are potent suppressors of HBV replication with high barriers to resistance. Pegylated interferon-alfa (Peg-IFN) is an alternative with finite treatment duration. Treatment aims for HBsAg loss (functional cure), viral suppression, and prevention of cirrhosis and HCC. Hepatitis B vaccination schedule: 3 doses (0/1/6 months) for susceptible adults; birth dose within 24 hours for neonates born to HBsAg-positive mothers (combined with HBIg for high-risk births). Hepatitis A vaccination: 2 doses (0/6–12 months) for travellers, food handlers, household contacts of HAV cases, patients with chronic liver disease. There is no vaccine for HCV or HEV (HEV-239 vaccine is available in China but not globally licensed).
Alcoholic liver disease: The cornerstone of management at every stage is complete abstinence from alcohol. For severe alcoholic hepatitis (DF >32), prednisolone 40 mg/day × 28 days is first-line in the absence of contraindications (infection, GI bleed, renal failure). Pentoxifylline has lost favour in recent trials. Nutritional support (high-calorie, high-protein diet) is essential as alcoholic patients are severely malnourished. Thiamine (vitamin B1) replacement is mandatory to prevent Wernicke's encephalopathy.
Complications of cirrhosis — management essentials:
- Ascites: Restrict dietary sodium (2 g/day); loop diuretic furosemide combined with spironolactone (aldosterone antagonist — targets the RAAS driving sodium retention); therapeutic paracentesis for refractory or tense ascites with albumin replacement (6–8 g per litre removed to prevent post-paracentesis circulatory dysfunction).
- SBP: Empirical third-generation cephalosporin (ceftriaxone or cefotaxime) IV for 5 days, started immediately when ascitic PMN ≥250/mm³; IV albumin 1.5 g/kg at diagnosis and 1 g/kg on day 3 reduces HRS development. Secondary prophylaxis: norfloxacin 400 mg/day indefinitely after a first SBP episode.
- Variceal haemorrhage: Acute: terlipressin (or somatostatin/octreotide) + endoscopic variceal ligation (EVL) or sclerotherapy; antibiotics (norfloxacin/ceftriaxone) to prevent SBP in the peri-bleed period. Secondary prevention: non-selective beta-blockers (propranolol/carvedilol) + repeat EVL sessions. TIPS (transjugular intrahepatic portosystemic shunt) for refractory bleeding.
- Hepatic encephalopathy: Treat precipitants first. Lactulose (produces colonic acidification → traps ammonia as ammonium; also a laxative to clear nitrogenous substrate) is first-line. Rifaximin (non-absorbable antibiotic reducing gut ammonia-producing bacteria) is added for recurrent or refractory HE. Protein restriction is no longer recommended (causes sarcopenia); aim for adequate protein intake.
- Hepatorenal syndrome: Terlipressin + IV albumin (20–40 g/day) is first-line; alternatively noradrenaline (ICU). TIPS as bridge to transplant. Dialysis if refractory. Liver transplantation is definitive.
- HCC screening: 6-monthly liver ultrasound ± AFP in all cirrhotic patients and high-risk HBV carriers. Treatment per BCLC stage.
- Liver transplantation: Indications include decompensated cirrhosis with MELD ≥15, HCC within Milan criteria (single nodule ≤5 cm or up to 3 nodules ≤3 cm each, no vascular invasion, no extrahepatic metastases), acute liver failure meeting King's College criteria. Contraindications: active substance abuse, extrahepatic malignancy, severe cardiopulmonary disease.
Self-Assessment: Integrating Liver Disease Pathophysiology
At this point in the module, you have covered the biochemical basis of hyperbilirubinaemia across its four mechanisms, the pathophysiology and clinical evolution of viral hepatitis (A–E), alcoholic liver disease from steatosis through alcoholic hepatitis to cirrhosis, the staged complications of portal hypertension with Child-Pugh and MELD scoring, SAAG interpretation, SBP diagnosis, HE West-Haven grading, HRS management, and HCC screening. You have also reviewed the diagnostic approach using LFT patterns and hepatitis serology sequences, and the management principles for each major condition. The clinical scenarios below are designed to consolidate and integrate this learning by requiring you to apply the full pathophysiology-to-management chain to realistic patient presentations. Work through each scenario before reading the analysis — this tests your ability to reason through, not merely recall.
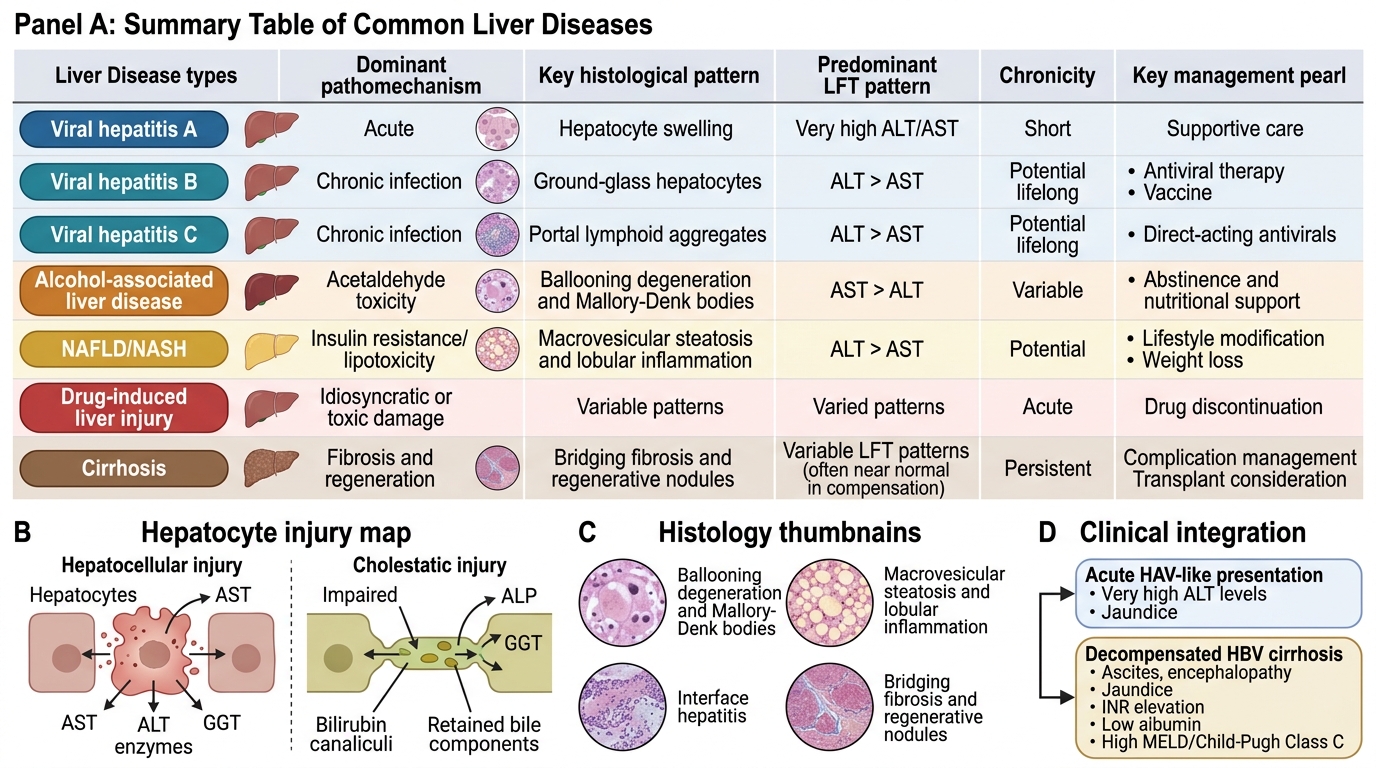
Integrated Summary of Major Liver Diseases
Scenario A: A 52-year-old man with known cirrhosis secondary to chronic HBV infection presents with increasing abdominal distension and confusion. Examination reveals ascites, asterixis, and caput medusae. Serum bilirubin is 5.1 mg/dL, albumin 2.0 g/dL, INR 2.8, creatinine 1.8 mg/dL. He has moderate ascites and grade II encephalopathy.
Self-check: Calculate his Child-Pugh score: bilirubin 5.1 mg/dL = 3 points; albumin 2.0 g/dL = 3 points; INR 2.8 = 3 points; ascites moderate = 2 points; HE grade II = 2 points. Total = 13 points → Child-Pugh Class C (decompensated cirrhosis). MELD = uses bilirubin, creatinine, INR (not albumin) — this patient has creatinine 1.8 (elevated) indicating renal involvement; MELD likely >20 suggesting significant short-term mortality. The pathophysiology chain: portal hypertension (HVPG >12 mmHg) → splanchnic vasodilatation → RAAS activation → ascites + sodium retention; hepatocellular failure → reduced ammonia clearance → HE grade II; jaundice from conjugation failure + intrahepatic cholestasis.
Scenario B: A 19-year-old student presents with 1 week of jaundice, nausea, and profound fatigue after returning from a rural area. ALT 1,620 IU/L, bilirubin 4.2 mg/dL (direct fraction 3.1 mg/dL). Anti-HAV IgM is positive.
Self-check: Acute hepatitis A. The markedly elevated ALT (hepatocellular pattern) reflects immune-mediated hepatocyte destruction. Direct bilirubin dominant = conjugated hyperbilirubinaemia from intrahepatic cholestasis in acute hepatitis (excretion impaired by hepatocyte injury). Management: supportive (fluids, rest, avoid alcohol/hepatotoxic drugs); no antiviral needed; counsel on faecal-oral precautions, vaccinate close contacts. No chronicity risk — HAV does not cause chronic infection. Warn about rare (<1%) fulminant hepatic failure, especially if underlying liver disease.
Scenario C: A 40-year-old woman who has been taking isoniazid for latent TB prophylaxis for 3 months develops nausea, anorexia, and jaundice. ALT 680 IU/L. She also takes paracetamol for chronic back pain at 3 g/day.
Self-check: DILI — two agents implicated. INH causes idiosyncratic hepatocellular DILI through reactive metabolite formation; paracetamol risk is amplified by INH because INH induces CYP2E1, increasing NAPQI production from paracetamol. Both agents must be stopped. RUCAM assessment needed. Liver function must be monitored to assess for resolution. If paracetamol hepatotoxicity is contributing, NAC should be considered. Key principle: both drugs withdrawn simultaneously, not one at a time.
CLINICAL PEARL
The SAAG is more reliable than total ascitic protein for categorising ascites because it corrects for plasma protein abnormalities common in cirrhosis. Always use the formula: SAAG = serum albumin − ascites albumin (not total protein). A SAAG ≥ 1.1 g/dL means portal hypertension is driving the ascites (cirrhosis in 85% of cases; also right heart failure, Budd-Chiari syndrome, constrictive pericarditis). A SAAG < 1.1 g/dL means the ascites is from a condition unrelated to portal hypertension (TB peritonitis, malignant ascites, pancreatitis, nephrotic syndrome). Never invert this: ≥1.1 = portal; <1.1 = non-portal.
A second pearl: Child-Pugh vs MELD — never cross-contaminate the parameters. Child-Pugh uses albumin + clinical ascites/encephalopathy (5 parameters). MELD uses creatinine + no albumin (3 laboratory values only). Creatinine does not appear in Child-Pugh; albumin does not appear in MELD. This is a frequent examination trap and a real clinical error.