Page 2 of 23
IM7.1-7 | Rheumatologic Disease Foundations — SDL Guide (Part 2)
Pathophysiology and Genetic Basis of Autoimmune Rheumatic Disease
Autoimmune rheumatic diseases are a group of conditions in which the immune system mounts a sustained, dysregulated attack against the body's own tissues — most prominently synovial joints, connective tissues, skin, kidneys, lungs, blood vessels, and the central nervous system. Understanding the mechanisms of autoimmunity is essential because it explains why DMARDs work, why biologics target specific cytokines, and why disease can flare and remit.
The central mechanism begins with failure of immune tolerance. In health, self-reactive T lymphocytes are eliminated in the thymus (central tolerance) and peripheral self-reactive cells are suppressed by regulatory T cells (Tregs), anergy, or activation-induced cell death. In autoimmune disease, multiple tolerance checkpoints fail simultaneously — driven by a combination of genetic susceptibility, environmental triggers, and stochastic immune dysregulation. The result is the activation of autoreactive T-helper (CD4+) cells and the loss of normal B-cell regulation, allowing the production of autoantibodies that bind self-antigens and trigger inflammation.
Genetic basis is polygenic and varies by disease. The strongest genetic associations are with the HLA (Human Leukocyte Antigen) system on chromosome 6, which encodes MHC molecules responsible for presenting peptide antigens to T cells. Different HLA haplotypes predispose to specific autoimmune conditions by determining which self-peptides are presented and whether autoreactive T-cell clones are deleted or activated:
- HLA-DR4 (specifically HLA-DRB104:01 and 04:04) — strongly associated with rheumatoid arthritis (RA), containing the shared epitope in the third hypervariable region
- HLA-B27 — associated with seronegative spondyloarthropathies (ankylosing spondylitis, reactive arthritis, psoriatic arthritis, IBD-associated arthritis); present in ~95% of ankylosing spondylitis patients vs ~8% of the general Indian population
- HLA-DR3 and DR2 (DQB10602) — associated with systemic lupus erythematosus (SLE)
- HLA-DR5, DR7 — associated with Sjögren syndrome and dermatomyositis
Beyond HLA, non-HLA genes contribute: PTPN22 (a protein tyrosine phosphatase gene) increases risk of RA and SLE; STAT4, IRF5 increase SLE risk; IL-23R mutations are associated with spondyloarthropathies. The interaction of multiple low-risk alleles rather than a single dominant mutation explains the incomplete penetrance and family clustering without Mendelian inheritance.
Environmental triggers are required to convert genetic susceptibility into overt disease. Key triggers include:
- Infection: molecular mimicry (bacterial peptides resembling self-antigens — e.g., Klebsiella pneumoniae nitrogenase and HLA-B27 in ankylosing spondylitis), bystander activation, and formation of NETs (Neutrophil Extracellular Traps) generating citrullinated proteins that drive RA
- Smoking: doubles the risk of RA, particularly in HLA-DR4 carriers; induces citrullination of proteins in lung tissue, triggering anti-CCP antibody production years before joint symptoms appear
- Ultraviolet radiation: activates keratinocytes to produce cytokines and triggers anti-nuclear antibody formation in SLE
- Hormonal factors: oestrogen augments immune responses, explaining the female predominance of SLE (9:1 F:M), RA (3:1), and Sjögren syndrome (9:1)
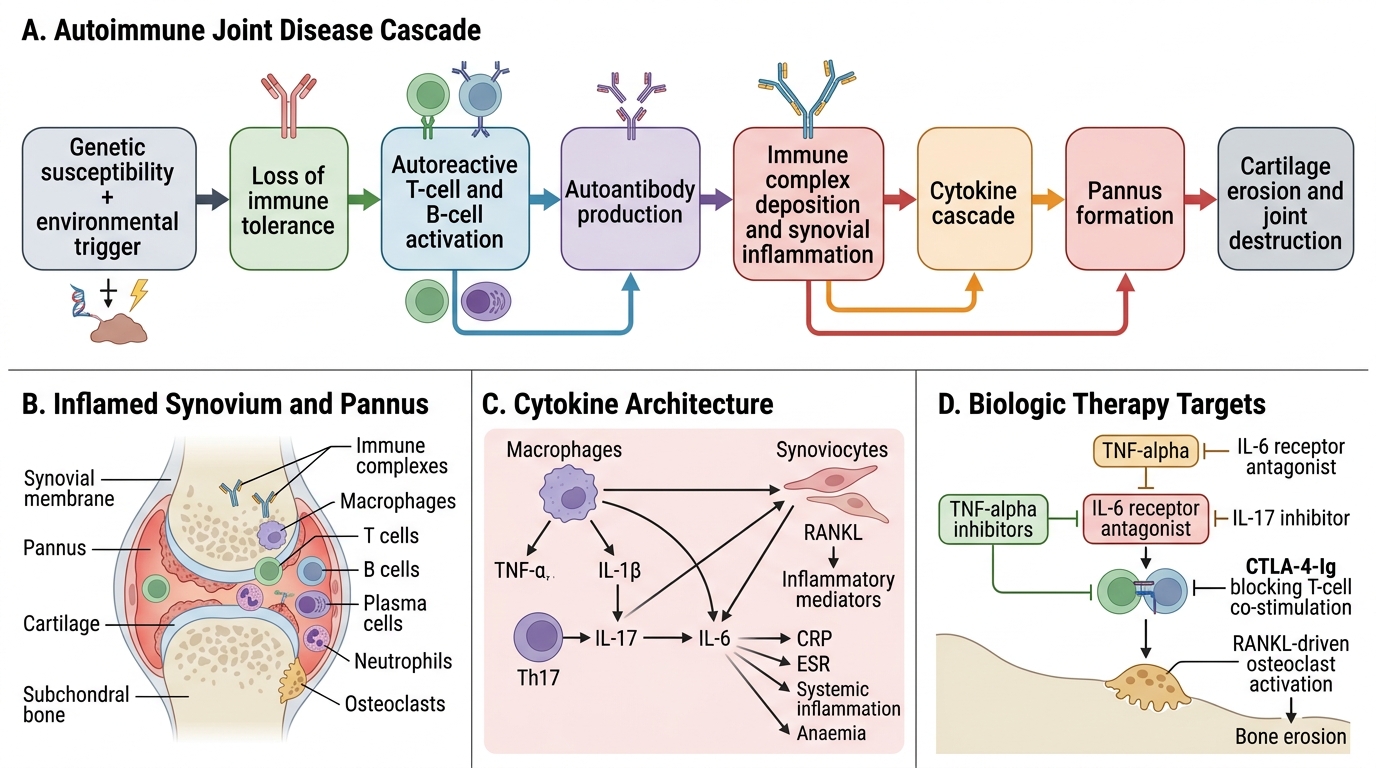
Pathophysiology of Autoimmune Joint Disease
Within the synovium, the effector phase of inflammation involves a cytokine cascade driven by macrophage-derived TNF-α and IL-1β, which activate synoviocytes to proliferate and form pannus — an invasive granulation tissue that erodes articular cartilage and subchondral bone. IL-6 drives acute-phase reactant production (CRP, ESR), systemic inflammation, and anaemia. IL-17 (from Th17 cells) promotes neutrophil recruitment and osteoclast activation. RANKL (Receptor Activator of Nuclear Factor κB Ligand) produced by activated T-cells and synoviocytes stimulates osteoclasts to resorb bone — producing the characteristic erosions seen on X-ray in RA. This cytokine architecture is directly targeted by biologic therapies: TNF-α inhibitors (etanercept, adalimumab, infliximab), IL-6 receptor antagonists (tocilizumab), IL-17 inhibitors (secukinumab), and CTLA-4-Ig (abatacept, which blocks T-cell co-stimulation).
Systemic Manifestations of Rheumatologic Disease
Rheumatic diseases are, at their core, systemic inflammatory conditions — the joint is often the most visible arena, but inflammation affects virtually every organ system. Recognising extra-articular and systemic manifestations is crucial both for diagnosis (skin, eye, and mucosal signs are often diagnostic clues that arrive before the joint picture is clear) and for monitoring disease activity and damage. A physician who only examines the joints and misses the malar rash of SLE, the iritis of ankylosing spondylitis, or the oral ulcers of Behçet disease has performed an incomplete assessment.
Constitutional features: fever (low-grade in RA, high-spiking quotidian fever with rash in adult-onset Still's disease — a classic fever pattern that should be specifically asked about), profound fatigue (a major quality-of-life issue in RA, SLE, and fibromyalgia), weight loss, anaemia of chronic disease (normocytic, normochromic, low reticulocytes, low serum iron with normal or elevated ferritin — a cytokine-mediated anaemia of inflammation), and lymphadenopathy (SLE, adult-onset Still's).
Skin manifestations:
- Malar (butterfly) rash: fixed erythema across the cheeks and nasal bridge, sparing the nasolabial folds — pathognomonic of SLE; photosensitive
- Discoid lupus: scarring, erythematous plaques with follicular plugging and central atrophy — can occur without systemic involvement
- Gottron's papules: violaceous papules over the MCP and IP joints — pathognomonic of dermatomyositis
- Heliotrope rash: purple discolouration of the upper eyelids — dermatomyositis
- Psoriasis: scaly erythematous plaques on extensor surfaces, scalp, umbilicus, intergluteal cleft — associated with psoriatic arthritis in ~20–30% of psoriasis patients; nail changes (pitting, onycholysis, oil-drop sign) are a clue
- Rheumatoid nodules: firm, non-tender subcutaneous nodules over pressure points (olecranon, dorsum of fingers, Achilles tendon) — seropositive RA
- Tophi (gout): chalky white deposits of monosodium urate crystals in the helix of the ear, finger pulps, olecranon bursa, and Achilles tendon — chronic tophaceous gout
- Raynaud's phenomenon: triphasic colour change of digits (white → blue → red) on cold exposure — primary (idiopathic) or secondary to systemic sclerosis, SLE, mixed connective tissue disease
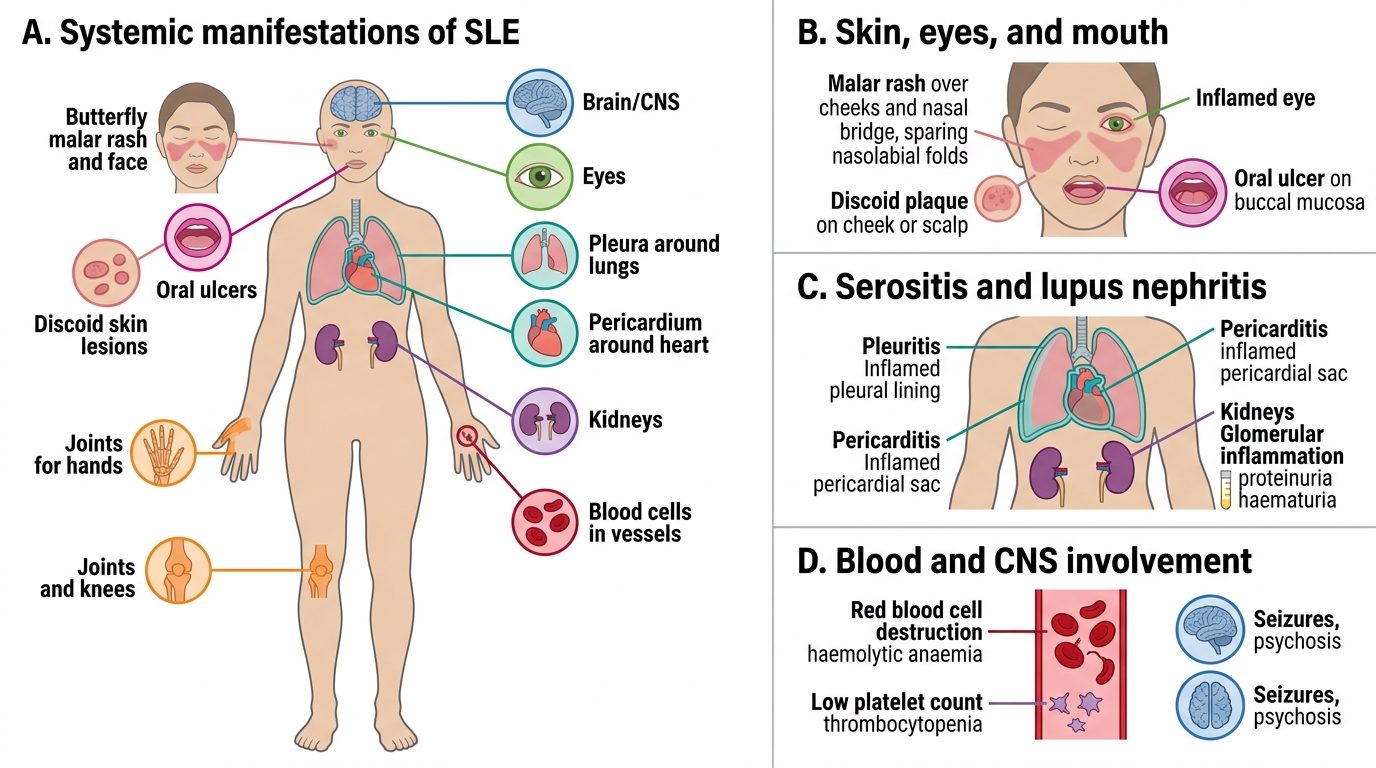
Systemic Manifestations of SLE
Eye manifestations:
- Anterior uveitis (iritis): acute red painful eye with photophobia — associated with HLA-B27 spondyloarthropathies. Requires urgent ophthalmological referral to prevent vision loss
- Episcleritis / scleritis: injection of sclera — RA, SLE, Wegener granulomatosis (GPA)
- Keratoconjunctivitis sicca (dry eyes): in Sjögren syndrome — assessed by Schirmer's test
- Band keratopathy: calcification of the cornea — juvenile idiopathic arthritis
Renal manifestations:
- Lupus nephritis: the most serious organ manifestation of SLE; classified ISN/RPS into 6 classes (Class I–VI); class III and IV = proliferative nephritis requiring immunosuppression (prednisolone + cyclophosphamide or mycophenolate mofetil); class V = membranous nephritis; urine dipstick for proteinuria and haematuria is mandatory in every SLE patient at every visit
- Renal amyloidosis (AA type): secondary to longstanding systemic inflammation in RA or AS
- ANCA-associated vasculitis: GPA and microscopic polyangiitis affect small renal vessels causing rapidly progressive glomerulonephritis
Pulmonary and cardiovascular manifestations:
- Interstitial lung disease (ILD): associated with RA, systemic sclerosis, polymyositis/dermatomyositis, Sjögren syndrome
- Pulmonary hypertension: a life-threatening complication of systemic sclerosis (CREST syndrome) and SLE
- Serositis (pleuritis/pericarditis): SLE, RA (RA pleuritis characteristically has very low pleural fluid glucose)
- Accelerated atherosclerosis: SLE and RA carry a 2–3-fold increased cardiovascular risk from chronic systemic inflammation
- Libman-Sacks endocarditis: non-infective verrucous vegetations on both sides of the mitral valve — SLE
- Aortitis and aortic regurgitation: ankylosing spondylitis
Neurological manifestations:
- Mononeuritis multiplex: asymmetric stepwise peripheral neuropathy — vasculitis
- Cervical myelopathy from atlantoaxial subluxation: a dangerous complication of RA requiring flexion-extension cervical X-rays before GA
- CNS lupus: seizures, psychosis, stroke-like episodes — associated with antiphospholipid antibodies
- Proximal myopathy: inflammatory myopathies — symmetric shoulder and hip girdle weakness, elevated CK
SELF-CHECK
A 28-year-old woman presents with a 3-month history of bilateral finger and wrist joint swelling with prolonged morning stiffness. She also has inflammatory-appearing swelling of the left 4th toe — the entire toe is swollen from base to tip. Which diagnosis does 'sausage digit' (dactylitis) most strongly suggest?
A. Rheumatoid arthritis
B. Systemic lupus erythematosus
C. Seronegative spondyloarthropathy (e.g., psoriatic arthritis)
D. Gout
Reveal Answer
Answer: C. Seronegative spondyloarthropathy (e.g., psoriatic arthritis)
Dactylitis ('sausage digit') — diffuse swelling of the entire digit including the flexor tendon sheath and all IP joints — is a hallmark of spondyloarthropathies, particularly psoriatic arthritis and reactive arthritis. It is not a feature of RA (which affects specific joints without tendon sheath involvement in this pattern), SLE (Jaccoud's arthropathy is reducible and non-erosive), or gout (which causes acute podagra at the first MTP joint, not dactylitis). The combination of hand/wrist synovitis with dactylitis should prompt examination for psoriatic skin/nail changes.
Diagnosis and Investigation of Rheumatologic Disease
A systematic clinical approach to joint pain integrates the pathophysiological classification framework into a structured clinical encounter. The goal is to arrive at a focused differential diagnosis before ordering investigations — not to use blood tests as a fishing expedition. The approach follows the format of a clinical encounter: history → examination → investigation → differential diagnosis. The key principle is that each investigation is ordered to test a specific hypothesis generated by the clinical assessment, not as a scatter-gun autoimmune panel.
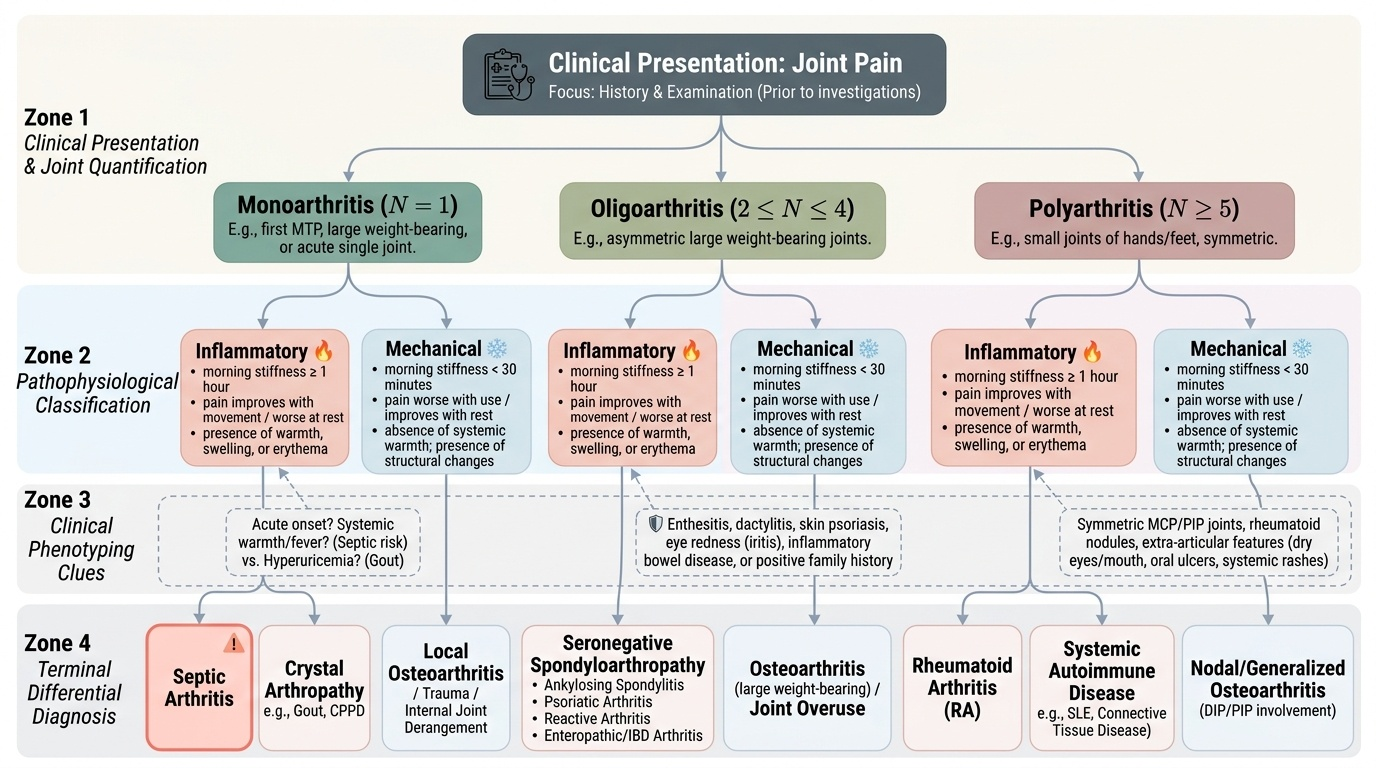
Provided image
Step 1 — Characterise the joint pain pattern. Ask: How many joints? (mono = 1; oligo = 2–4; poly = ≥5). Which joints? (small hands/feet = RA; large weight-bearing = OA, seronegative spondyloarthropathy, septic; first MTP = gout; axial = AS). Symmetric or asymmetric? (symmetric poly = RA, symmetric OA; asymmetric oligo = seronegative; migratory = rheumatic fever, gonococcal arthritis). Acute or chronic?
Step 2 — Determine inflammatory vs mechanical. Duration of morning stiffness (≥1 hour = inflammatory; <30 min = mechanical). Pain at rest vs with use. Presence of warmth, swelling, systemic features.
Step 3 — Identify the anatomical source. Is the pain articular or periarticular? Is there enthesitis (spondyloarthropathy)? Dactylitis?
Step 4 — Screen for extra-articular/systemic manifestations. Skin (rash, psoriasis, tophi, nodules), eyes (iritis, dry eyes), mouth (oral ulcers — Behçet, SLE, reactive arthritis), genitourinary history (urethritis/cervicitis + arthritis + conjunctivitis = Reiter triad), bowel symptoms (IBD-associated arthritis), family history of psoriasis, AS, or IBD.
Step 5 — Direct investigations efficiently. CBC + ESR + CRP (inflammation marker), RF + anti-CCP (RA), ANA + anti-dsDNA (SLE), HLA-B27 (spondyloarthropathy), uric acid + synovial fluid crystal analysis (gout/pseudogout), synovial fluid culture (septic arthritis), X-rays of affected joints (erosions, joint space narrowing, osteophytes), DEXA (bone density in chronic inflammatory disease on steroids). In India, always consider TB joint disease in any chronic monoarthritis — weight-bearing joints with a chronic insidious course warrant Mantoux test and synovial biopsy for AFB.
SELF-CHECK
A 22-year-old man presents with a 4-week history of lower back pain (worse at night and in the morning, improving with exercise), right knee swelling, and painful red eye (anterior uveitis confirmed by ophthalmology). He reports urethral discharge 6 weeks prior. What is the most likely diagnosis?
A. Rheumatoid arthritis
B. Ankylosing spondylitis
C. Reactive arthritis (formerly Reiter syndrome)
D. Gonococcal arthritis
Reveal Answer
Answer: C. Reactive arthritis (formerly Reiter syndrome)
The triad of arthritis (asymmetric oligoarthritis — knee), anterior uveitis, and preceding urogenital infection (urethral discharge) constitutes reactive arthritis (formerly Reiter syndrome). The classic triad is 'can't see, can't pee, can't climb a tree' — conjunctivitis/uveitis, urethritis/cervicitis, arthritis. Inflammatory back pain (worse at rest/morning, improving with exercise) indicates axial involvement. HLA-B27 is positive in ~75% of cases. Ankylosing spondylitis is a chronic axial spondyloarthropathy without the genitourinary trigger. RA is symmetric small joint polyarthritis. Gonococcal arthritis is typically migratory polyarthralgia with skin pustules in a sexually active young person.