Page 27 of 53
PE19.10 | Neonatal Hypoglycemia — SDL Guide
Learning Objectives
- Explain neonatal glucose physiology and temperature regulation in the newborn transition period
- Identify at-risk neonates for hypoglycemia and describe screening protocols
- Describe the aetiology and pathophysiology of neonatal hypoglycemia
- Recognise the clinical features of neonatal hypoglycemia including asymptomatic presentations
- Outline the stepwise management using oral feeding, 10% dextrose bolus, and glucose infusion rate
INSTRUCTIONS
Neonatal hypoglycemia is one of the most common and potentially serious metabolic emergencies in the newborn period. Failure to recognise and treat low blood glucose promptly can cause permanent neurological damage including intellectual disability, seizure disorders, and cerebral palsy. As a final-year student, you must be able to identify at-risk neonates, interpret bedside glucose values, and initiate the correct management — particularly the weight-based 10% dextrose bolus, not the adult 50% dextrose that causes rebound hypoglycemia and vascular injury in neonates.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 7 — Neonatal Metabolic Disorders (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 125 — Hypoglycemia (textbook)
- NNF (National Neonatology Forum) India — Clinical Practice Guidelines on Neonatal Hypoglycemia (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 2.1 kg baby is born at 36 weeks of gestation to a mother with gestational diabetes. Twelve hours after birth, the nurse finds the baby jittery and feeding poorly. A bedside glucometer reads 32 mg/dL. The on-call intern reaches for a syringe of 50% dextrose — the same she uses for adult hypoglycemia. Should she use it? What is the correct weight-based dose and concentration for this neonate, and what should come immediately after?
WHY THIS MATTERS
Neonatal hypoglycemia (blood glucose below 45 mg/dL) affects 1–3 per 1,000 live births overall but rises to 10–15% among at-risk groups such as infants of diabetic mothers, small-for-gestational-age neonates, and preterm babies. The neonatal brain depends almost entirely on glucose for energy, and even brief symptomatic episodes can cause hippocampal injury, periventricular white-matter damage, and long-term neurodevelopmental sequelae. Early identification through systematic screening and correct weight-based treatment are therefore core clinical competencies. Understanding neonatal glucose physiology explains why the transition from intrauterine to extrauterine life is a critical window, and why certain conditions predictably disrupt this transition.
RECALL
From your Physiology training, recall that blood glucose is maintained by the balance between glycogenolysis (breakdown of hepatic glycogen) and gluconeogenesis (synthesis of glucose from lactate, glycerol, and amino acids), regulated by the counter-regulatory hormones glucagon, adrenaline, cortisol, and growth hormone. Insulin, secreted by pancreatic β-cells in response to hyperglycaemia, drives glucose uptake and glycogen synthesis. In utero, the fetus receives continuous glucose from the mother across the placenta. This constant supply stimulates fetal β-cell hyperplasia and keeps fetal insulin levels high. At birth, the umbilical cord is cut and the glucose supply drops abruptly — demanding an immediate counter-regulatory response. From neonatal physiology, recall that brown adipose tissue (BAT) provides non-shivering thermogenesis in response to cold stress; this thermogenic metabolic demand consumes additional glucose and free fatty acids, further stressing glucose homeostasis in the immediate newborn period.
Neonatal Glucose Physiology and Temperature Regulation
At birth, the neonate's blood glucose drops from the maternal level (~80–90 mg/dL) to a physiological nadir of approximately 30–40 mg/dL within the first 1–2 hours of life. This early dip is normal and transient, triggered by the abrupt cessation of placental glucose transfer and the spike of counter-regulatory hormones — principally glucagon and catecholamines — that activate glycogenolysis and gluconeogenesis. In healthy term neonates, glucose stabilises above 45 mg/dL by 2–4 hours, sustained by hepatic glycogen (which accumulates during the third trimester), initiation of enteral feeding, and ketone production from fat oxidation as an alternative fuel.
Temperature regulation is tightly coupled to glucose metabolism in neonates. Neonates have a large surface-area-to-body-mass ratio, thin skin with minimal subcutaneous insulation, and limited capacity for shivering. They rely on non-shivering thermogenesis — the oxidation of stored triglycerides in brown adipose tissue (BAT), which is distributed in the nape of the neck, interscapular region, axillae, and perinephric area. This BAT-mediated heat generation consumes significant glucose and fatty acids. Cold stress therefore accelerates glucose consumption and depletes stores rapidly, creating a direct metabolic link between thermal instability and hypoglycemia. The neutral thermal environment (NTE) — the ambient temperature at which oxygen and glucose consumption are minimised — is approximately 32–34°C for term neonates and 34–36°C for preterm neonates weighing less than 1,500 g. Keeping neonates warm reduces their caloric demand and is itself a preventive intervention for hypoglycemia.
This dual dependency — on hepatic substrate stores AND on thermal stability — explains why preterm neonates, who are born before the third-trimester accumulation of hepatic glycogen and BAT is complete, are particularly vulnerable to hypoglycemia even without any additional metabolic disorder.
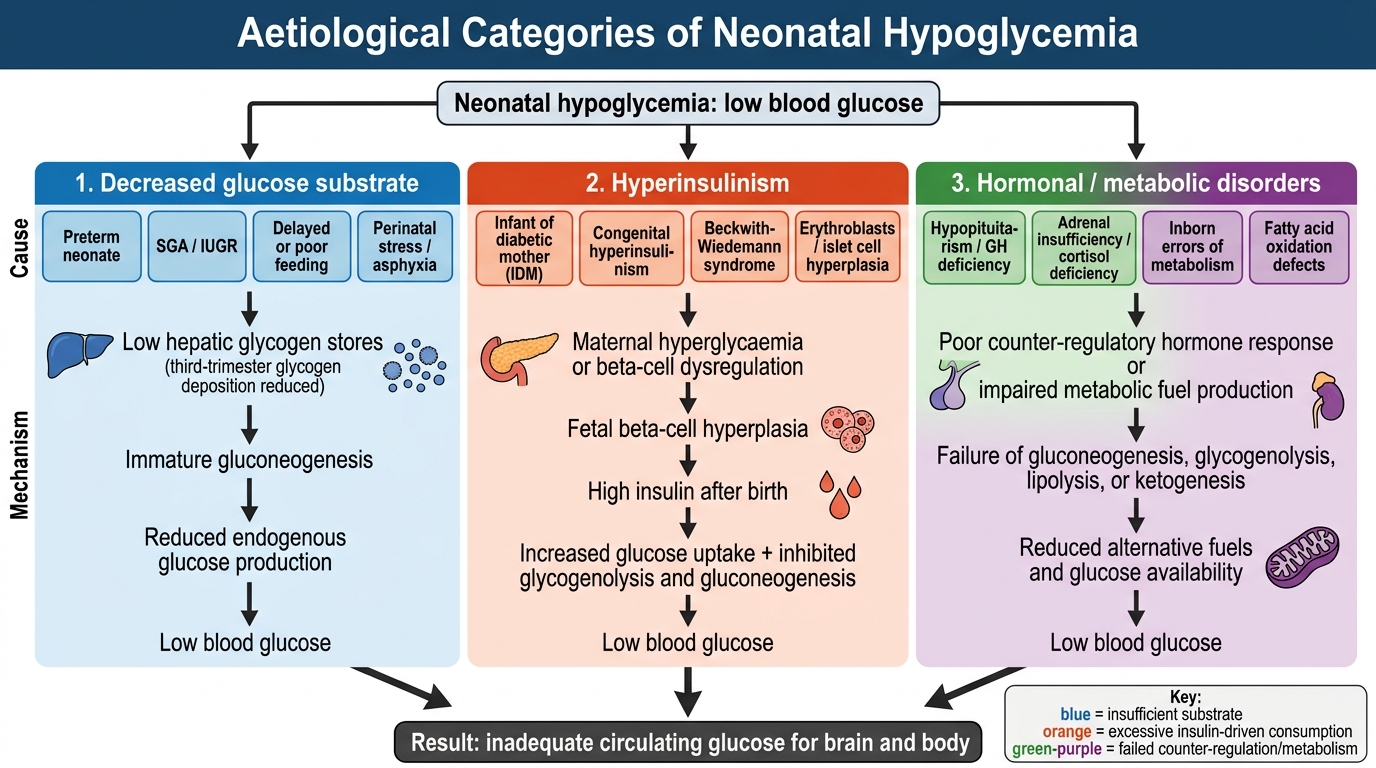
Aetiology and Pathophysiology of Neonatal Hypoglycemia
Pathophysiology and Aetiology of Neonatal Hypoglycemia
Neonatal hypoglycemia arises from three broad pathophysiological mechanisms: insufficient glucose substrate, excessive glucose consumption (often driven by hyperinsulinism), or a failure of counter-regulatory hormone responses. Understanding which mechanism is operating guides both the urgency and the escalation of treatment.
1. Decreased glucose substrate is the most common cause in preterm and small-for-gestational-age (SGA) neonates. Hepatic glycogen is deposited during the third trimester (peak accumulation at 36–40 weeks). A preterm neonate born at 32–34 weeks has only 30–50% of term glycogen stores. An SGA or IUGR (intrauterine growth-restricted) neonate has suffered chronic placental insufficiency; hepatic glycogen stores are depleted, and liver mass itself is reduced. Gluconeogenic capacity is also immature in both groups.
2. Hyperinsulinism is the most common cause of severe and persistent hypoglycemia. The infant of a diabetic mother (IDM) exemplifies transient hyperinsulinism: maternal hyperglycaemia drives fetal hyperglycaemia → fetal β-cell hyperplasia → elevated fetal insulin. After birth, maternal glucose supply stops but insulin remains elevated, driving glucose into fetal tissues and suppressing ketogenesis. This explains why IDM hypoglycemia typically presents within 1–2 hours of birth and requires IV dextrose. Beckwith-Wiedemann syndrome (macrosomia, macroglossia, omphalocele) and congenital hyperinsulinism (mutations in KATP channel subunits SUR1/Kir6.2) cause persistent hyperinsulinism refractory to oral feeds.
3. Other causes include polycythaemia (haematocrit >65%; increased glucose consumption by excess erythrocytes), neonatal sepsis (increased metabolic demand + impaired gluconeogenesis + adrenal insufficiency), perinatal asphyxia (anaerobic glycolysis depletes glycogen; corticosteroid release impaired), inborn errors of metabolism (galactosaemia, fatty acid oxidation defects, glycogen storage diseases — rare but important), and endocrine deficiencies (adrenal insufficiency, growth hormone deficiency, panhypopituitarism).
| Cause | Onset | Key feature |
|---|---|---|
| IDM / LGA | First 1–6 h | Macrosomic; transient; responds to feeds |
| Preterm | Any time first 48 h | Low stores; concurrent respiratory issues |
| SGA/IUGR | First 12–24 h | Wasted; glycogen-depleted |
| Polycythaemia | First 12 h | Plethoric; haematocrit >65% |
| Sepsis | Any time | Unwell; other sepsis signs |
| Congenital hyperinsulinism | After 24 h–weeks | Persistent; diazoxide-responsive or surgical |
| Galactosaemia | After milk feeds | Jaundice, hepatomegaly, E. coli sepsis |
Clinical Features and Recognition
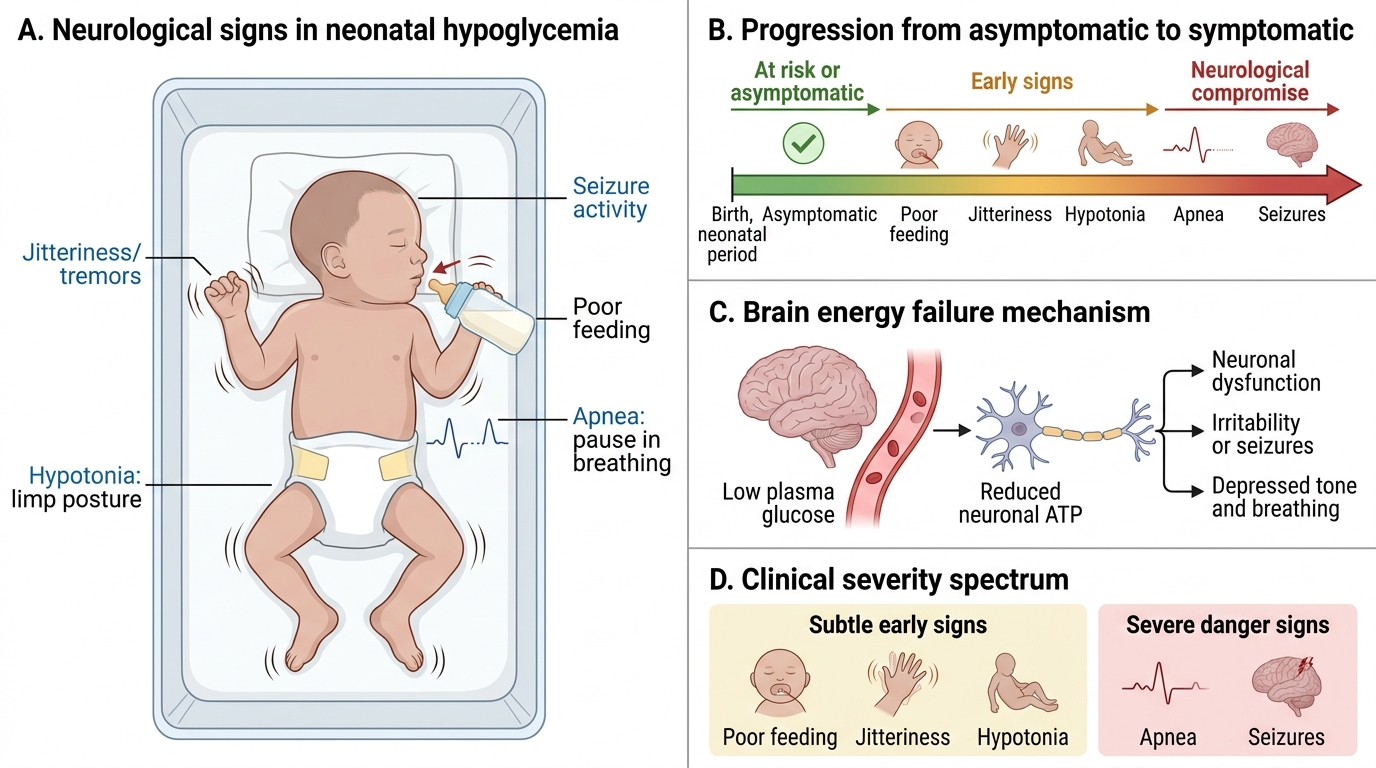
Neonatal hypoglycemia exists on a spectrum from entirely asymptomatic to life-threatening neuroglycopenia. The distinction between asymptomatic and symptomatic hypoglycemia is clinically important because it influences treatment urgency, route of administration, and monitoring intensity. Critically, the absence of symptoms does not exclude significant hypoglycemia — neonates can have blood glucose as low as 20–25 mg/dL without obvious signs, yet be sustaining neuronal injury. The neonatal brain, unlike the adult brain, has limited capacity to use ketone bodies as an alternative fuel substrate in the acute phase, making it exquisitely sensitive to glucose deprivation. This is why asymptomatic hypoglycemia in high-risk neonates — particularly those with hyperinsulinism — can cause white-matter injury without any overt clinical warning. The practitioner's role is therefore to detect hypoglycemia before it becomes symptomatic.
Asymptomatic hypoglycemia is detected only by routine screening of at-risk neonates. It accounts for the majority of cases and is managed with enteral feeds in milder instances.
Symptomatic hypoglycemia presents with non-specific neurological and autonomic signs:
- Jitteriness and tremors — the most common presenting symptom; coarse, repetitive trembling of limbs, easily triggered by handling
- Hypotonia — generalised floppiness; reduced spontaneous movement
- Poor feeding and weak suck — early sign, often misattributed to sleepiness
- Apnoea — central respiratory pauses, particularly in preterm neonates
- Cyanosis — may accompany apnoea or cardiovascular compromise
- High-pitched cry — neurological irritability
- Seizures — focal or multifocal clonic; tonic posturing; a late, serious sign indicating neuroglycopenia
- Lethargy progressing to coma — severe prolonged hypoglycemia
Jitteriness must be distinguished from seizures: jitteriness is stimulus-sensitive (can be stopped by gentle restraint of the limb), not accompanied by gaze abnormalities, and EEG is normal; seizures are stimulus-insensitive, may have eye deviation, and require EEG confirmation.
Neurological Signs of Neonatal Hypoglycemia