Page 11 of 30
PE20.4 | Hematuria — SDL Guide (Part 2)
Management and Referral Criteria
Management of haematuria in children is directed at the underlying cause, and the first management decision — whether to treat, observe, or refer — follows directly from the glomerular versus non-glomerular classification and the presence or absence of red-flag features. The most important management principle is that isolated asymptomatic microscopic haematuria with isomorphic RBCs, normal blood pressure, normal renal function, and no proteinuria is usually benign and does not require immediate invasive investigation or referral — but it does require careful periodic monitoring because a small proportion will unmask progressive glomerular disease on follow-up. The identification of red flags — which indicate a higher probability of significant glomerular disease, hereditary nephropathy, urological pathology, or systemic disease — determines who needs urgent specialist review.
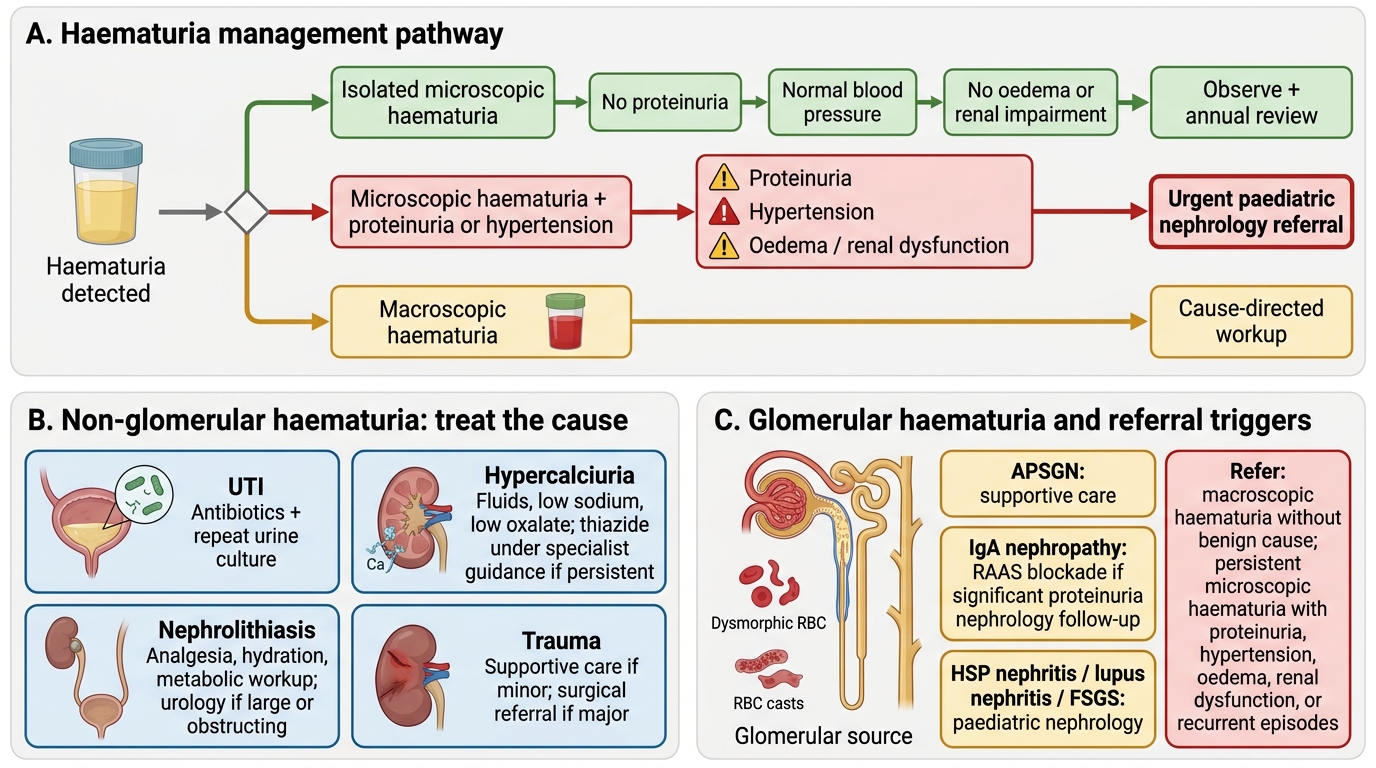
Management Pathways for Haematuria in Children
Non-glomerular haematuria — treat the cause:
• UTI: appropriate antibiotic course (per PE20.1 management); repeat urine culture at the end of treatment to confirm eradication.
• Hypercalciuria: increase fluid intake (first-line); reduce dietary sodium (sodium loading increases urinary calcium excretion); reduce oxalate-rich foods. If symptomatic hypercalciuria with or without calculi persists despite dietary measures, hydrochlorothiazide (reduces urinary calcium) may be used under specialist guidance.
• Nephrolithiasis: analgesia, hydration, dietary modification; metabolic workup for the stone type; urological referral for large or obstructing stones.
• Trauma: observation and supportive care for minor renal trauma; surgical referral for major injury.
Glomerular haematuria:
• APSGN: supportive management as covered in PE20.2 (penicillin, antihypertensives, diuretics).
• IgA nephropathy: no proven disease-modifying therapy for mild IgA nephropathy; RAAS blockade (ACE inhibitor or ARB) if significant proteinuria develops; nephrology follow-up.
• Other glomerular causes (HSP nephritis, lupus nephritis, FSGS): managed by paediatric nephrology.
Referral criteria to paediatric nephrology:
• Macroscopic haematuria without an identified benign cause
• Persistent microscopic haematuria (≥3 confirmations over ≥3 months) with any of the following: proteinuria (UPCR >0.2), hypertension, family history of CKD/ESRD, sensorineural deafness
• Haematuria + nephrotic-range proteinuria (any age)
• Haematuria + hypertension + renal impairment
• Low serum C3 persisting beyond 8–12 weeks
• Suspected hereditary nephritis (Alport syndrome: haematuria + family history + deafness)
• Suspected Wilms tumour or other renal mass (urgent surgical oncology referral)
• Haematuria in the context of systemic disease (HSP, lupus, vasculitis)
Self-Assessment
Haematuria is a presenting complaint that tests clinical reasoning across several simultaneous dimensions: Is the dipstick genuinely detecting blood? Is the blood coming from the glomerulus or the urological tract? If glomerular, which nephritis pattern does the clinical and laboratory picture suggest? If non-glomerular, is the cause benign (hypercalciuria, UTI) or serious (Wilms tumour, trauma)? Does this child need reassurance, treatment of a non-glomerular cause, a structured monitoring plan, or urgent nephrology referral? Answering these questions sequentially — rather than jumping to investigation or referral — is the mark of sound clinical reasoning. Before attempting the questions below, consolidate the glomerular/non-glomerular distinction (the pivot of the whole module), the RBC morphology findings that operationalise it, and the specific red-flag features that mandate specialist referral.
- Haematuria confirmed by ≥5 RBC/hpf on microscopy (≥5 RBC/mm³ unspun); dipstick alone is insufficient.
- Dysmorphic RBCs (acanthocytes) + RBC casts = glomerular origin.
- Isomorphic RBCs, no casts = non-glomerular origin.
- IgA nephropathy: synpharyngitic (no latent period), C3 normal; vs APSGN: latent period 1–2 weeks, C3 low.
- Hypercalciuria: commonest non-glomerular cause of isolated microscopic haematuria; spot urine Ca/Cr >0.2.
- Alport syndrome: X-linked COL4A5 mutation; haematuria + progressive proteinuria + sensorineural deafness + anterior lenticonus.
- Referral triggers: proteinuria + haematuria, hypertension + haematuria, persistent >3 months, family history CKD, low C3 beyond 8–12 weeks, macroscopic without identified cause.
SELF-CHECK
A 7-year-old girl has persistent microscopic haematuria confirmed on three occasions over 4 months. Her blood pressure is normal, UPCR is 0.15, serum albumin and creatinine are normal, C3 is normal, urine culture is negative, and USG KUB is normal. Her mother also has microscopic haematuria but normal renal function. What is the MOST likely diagnosis?
A. IgA nephropathy
B. Alport syndrome
C. Thin glomerular basement membrane (thin GBM) disease / benign familial haematuria
D. Hypercalciuria
Reveal Answer
Answer: C. Thin glomerular basement membrane (thin GBM) disease / benign familial haematuria
This presentation is classic for thin GBM disease (benign familial haematuria): isolated persistent microscopic haematuria, no proteinuria, no hypertension, normal renal function, normal C3, and family history of haematuria (autosomal dominant inheritance, typically heterozygous COL4A3/A4 mutations). Alport syndrome typically has more aggressive disease in males (progressive proteinuria, renal impairment, sensorineural deafness, anterior lenticonus). IgA nephropathy more often presents with episodic macroscopic haematuria. Hypercalciuria is excluded by the normal USG and normal urine Ca/Cr (would have been checked in non-glomerular workup).