Page 4 of 20
PE21.2 | Henoch Schonlein Purpura — SDL Guide
Learning Objectives
- Describe the classical tetrad of clinical features in Henoch Schönlein Purpura (IgA vasculitis)
- Explain the pathogenesis of HSP including aberrant IgA1 glycosylation and leukocytoclastic vasculitis
- Apply the EULAR/PRINTO/PRES 2010 classification criteria to diagnose HSP
- Select appropriate investigations including renal assessment and interpret ISKDC renal histological grading
- Outline the management of HSP from supportive care to immunosuppression for severe nephritis, and plan appropriate follow-up
INSTRUCTIONS
Henoch Schönlein Purpura — now formally named IgA vasculitis — is the most common systemic vasculitis in childhood. It strikes children between 3 and 10 years, usually following an upper respiratory infection, and produces a striking clinical tetrad that is highly recognisable once you have seen it. Most cases are self-limiting and resolve within 4–6 weeks, but the renal complication can be insidious and long-lasting. Understanding when HSP is benign and when it needs escalation — particularly its renal monitoring — is a core competency for every paediatrician. This module takes you from first clinical encounter to safe discharge and follow-up.
References
- Ghai Essential Pediatrics, 9th Ed, Ch 17 — Rheumatic Diseases (textbook)
- Nelson Textbook of Pediatrics, 21st Ed, Ch 182 — Henoch-Schönlein Purpura (textbook)
- EULAR/PRINTO/PRES 2010 Classification Criteria for HSP (IgA Vasculitis) (guideline)
- SHARE Guidelines for HSP Management in Children, 2019 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 6-year-old boy is brought to the emergency department with a 2-day history of a rash on both legs and buttocks, colicky abdominal pain, and difficulty walking due to painful swollen ankles. His parents noticed the rash after he recovered from a sore throat 10 days ago. On examination he has striking non-blanching, palpable, red-purple spots and patches on both lower limbs and buttocks. His platelet count is 240 × 10³/µL (normal), and urine dipstick shows 2+ blood and 1+ protein. He is afebrile and his abdomen is mildly tender. What is your diagnosis, and what will you do about the blood in his urine?
WHY THIS MATTERS
Henoch Schönlein Purpura (HSP), now classified as IgA vasculitis in the 2012 International Chapel Hill Consensus Conference nomenclature, is the most common systemic vasculitis in childhood. It affects predominantly children aged 3–10 years, with a slight male predominance. While the majority of cases resolve spontaneously within 4–6 weeks, up to 60% develop some degree of renal involvement, and a small subset progress to chronic kidney disease. The challenge is that renal involvement can appear weeks after the initial purpuric rash and may worsen silently — meaning the initial reassuring resolution of skin and joint symptoms does not signal that follow-up can stop. Every paediatrician must know the monitoring schedule and the triggers for escalation.
RECALL
Before proceeding, activate your prior knowledge. Recall from Physiology: IgA structure (IgA1 vs IgA2 subclasses), the role of IgA in mucosal immunity, and complement activation via the lectin and alternative pathways. Recall from Anatomy: the layers of the small-vessel wall (endothelium, basement membrane, tunica media) and the concept of immune complex deposition in vessel walls. Recall from your renal studies: the ISKDC classification of glomerulonephritis and the significance of haematuria and proteinuria as markers of renal injury. These will be directly relevant when you study the pathogenesis and renal complication of HSP.
Clinical Presentation of HSP
The clinical presentation of HSP is characterised by a tetrad of features that appear in a fairly predictable sequence, although not all features need to be present simultaneously. The most striking and diagnostically essential feature is the rash, which is typically preceded by an upper respiratory tract infection 1–3 weeks earlier.
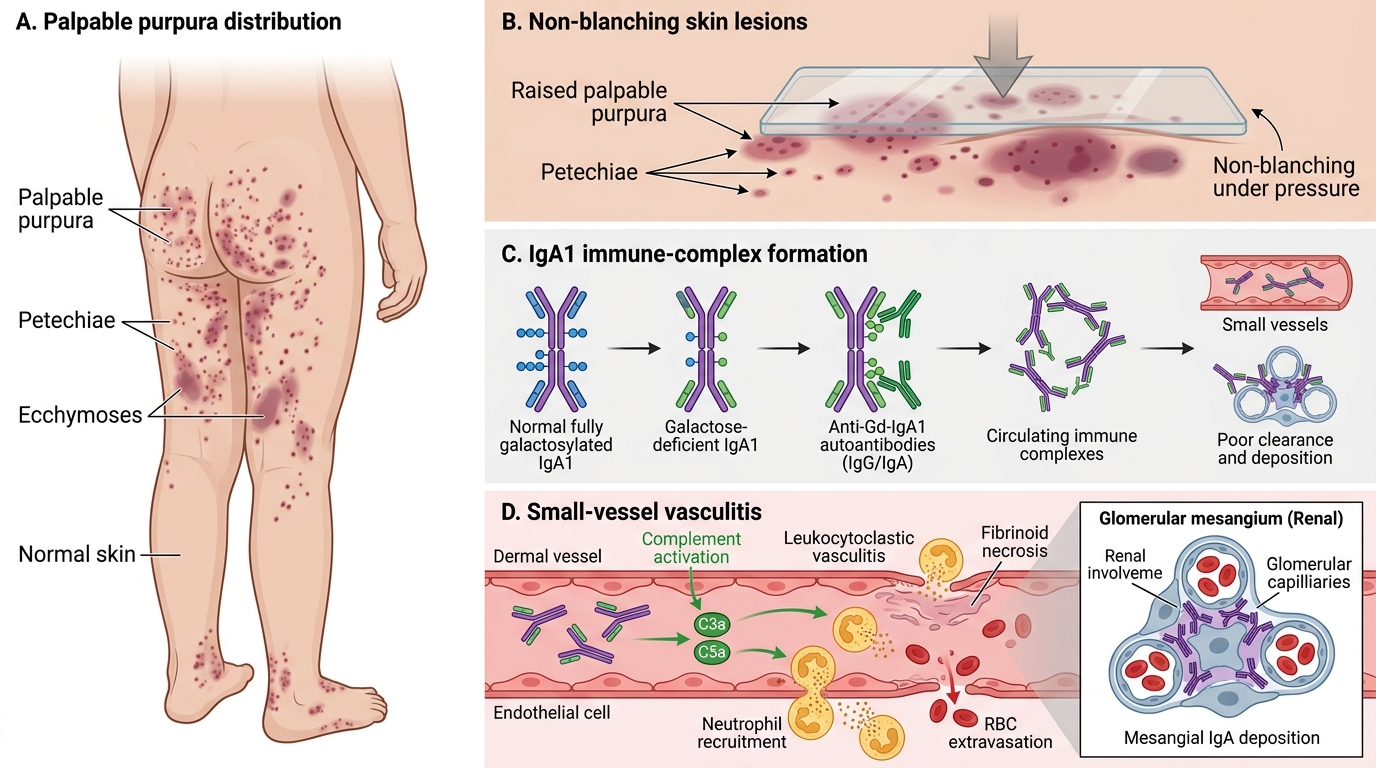
Palpable purpura is the mandatory diagnostic criterion and the hallmark of HSP. Unlike the petechiae of thrombocytopenia (which are flat and caused by platelet deficiency), the purpura of HSP is palpable — it stands slightly above the skin surface because the underlying inflammation causes vascular leakage and perivascular inflammatory cell infiltration. The lesions are distributed symmetrically over the buttocks and lower limbs (dependent areas and areas subject to pressure), ranging from small petechiae to large ecchymoses. Critically, the platelet count is normal — the purpura is vasculitic, not thrombocytopenic. On blanching, the lesions do not disappear, distinguishing them from haemangiomas or erythema.
Arthritis or arthralgia occurs in 60–80% of children and typically affects large joints of the lower limbs — knees and ankles. The joint involvement is periarticular (periarthritis) rather than true synovitis in most cases; it is painful and can cause limping but does not cause permanent joint damage. It usually resolves within days to weeks.
Abdominal pain occurs in 50–70% and results from submucosal and subserosal haemorrhage and oedema in the bowel wall. The pain is colicky and may be severe. Serious gastrointestinal complications include intussusception (1–5% — typically ileo-ileal), gastrointestinal bleeding, and, rarely, bowel ischaemia or perforation. Occult blood in stool is common. Abdominal ultrasound is essential if pain is severe or features of intussusception are present (colicky pain + vomiting + sausage-shaped mass).
Renal involvement (HSP nephritis) occurs in 20–60% of children. It typically manifests as microscopic haematuria and/or proteinuria, appearing within the first 4–8 weeks of disease but occasionally as late as 6 months after the rash. Most cases are mild and resolve, but 1–3% progress to end-stage renal disease. Macroscopic haematuria, nephrotic-range proteinuria, hypertension, or renal impairment are markers of severe nephritis.
Other features: scrotal oedema and pain (in boys, from vasculitis of scrotal vessels — important to distinguish from testicular torsion by Doppler ultrasound), CNS involvement (seizures, headache — rare), and pulmonary haemorrhage (very rare).
Henoch-Schonlein Purpura: Palpable Purpura and IgA Vasculitis
Pathophysiology and Aetiology of HSP
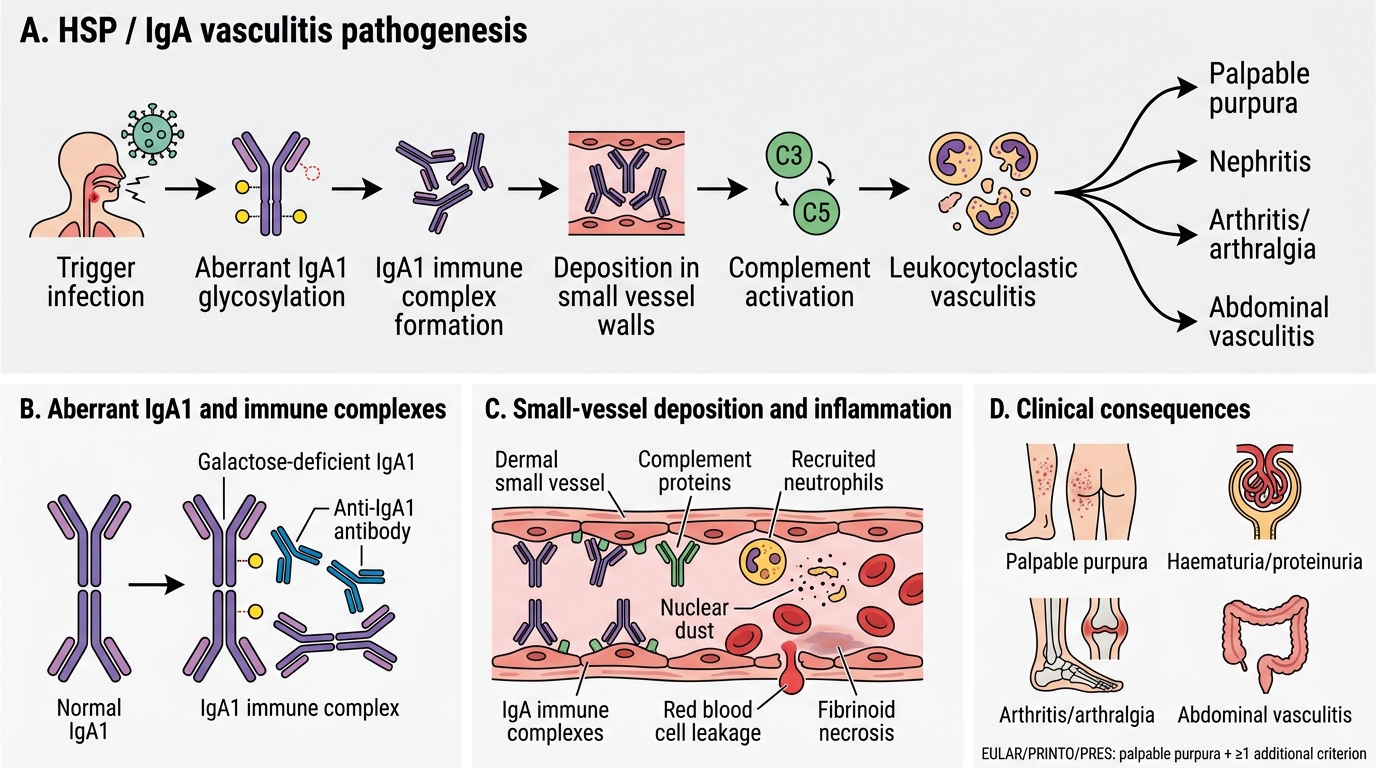
The central pathological event in HSP is the deposition of IgA1-containing immune complexes in small vessel walls, triggering complement activation and a leukocytoclastic vasculitic response. Understanding this mechanism explains the multi-system involvement, the triggers, and the rationale for immunosuppression in severe cases.
The aetiopathogenesis begins with an abnormality of IgA1 glycosylation. In healthy individuals, IgA1 (the predominant serum IgA subclass) has O-linked oligosaccharides on its hinge region that are fully galactosylated. In children with HSP, these oligosaccharides are aberrantly galactose-deficient (Gd-IgA1), exposing core GalNAc residues. These galactose-deficient IgA1 molecules are recognised as neo-antigens by IgG or IgA auto-antibodies, forming immune complexes (Gd-IgA1 + anti-Gd-IgA1). These large immune complexes are poorly cleared by the liver and accumulate in circulation, then deposit in the mesangium of glomeruli (causing IgA nephropathy pattern) and in the walls of small vessels (arterioles, capillaries, venules) throughout the body.
In vessel walls, deposited IgA1 complexes activate complement via the lectin and alternative pathways, generating C3a and C5a (anaphylatoxins). These attract and activate neutrophils, which infiltrate the vessel wall, degranulate, and fragment (karyorrhexis), releasing reactive oxygen species and proteases that cause fibrinoid necrosis of the vessel wall. This is the histological pattern of leukocytoclastic vasculitis — neutrophil infiltration, nuclear dust (fragmented nuclei), and fibrinoid necrosis with IgA deposits on direct immunofluorescence. The resulting vascular damage causes the leakage and perivascular haemorrhage that produces the palpable purpura in skin, haemorrhage and oedema in the bowel wall (abdominal pain), periarthritis (joint symptoms), and glomerular injury (nephritis).
Triggers: HSP is frequently preceded by upper respiratory tract infections, most commonly Streptococcus pyogenes (Group A Streptococcus), but also other bacteria and viruses (adenovirus, parvovirus B19, EBV). Other triggers include drugs, food, insect bites, and vaccinations. The infection is thought to drive increased mucosal IgA production, amplifying circulating levels of Gd-IgA1 complexes.
Pathogenesis of HSP / IgA Vasculitis
SELF-CHECK
A 7-year-old girl presents with palpable purpura on her legs and buttocks, ankle pain, and 3+ blood on urine dipstick. Her platelet count is 230 × 10³/µL and coagulation screen is normal. Skin biopsy shows leukocytoclastic vasculitis with IgA deposits on immunofluorescence. Which criterion from the EULAR/PRINTO/PRES 2010 classification does the biopsy finding satisfy?
A. The mandatory palpable purpura criterion
B. The renal involvement criterion
C. The IgA biopsy criterion (one of the four additional criteria)
D. The arthritis/arthralgia criterion
Reveal Answer
Answer: C. The IgA biopsy criterion (one of the four additional criteria)
The EULAR/PRINTO/PRES 2010 criteria for HSP/IgA vasculitis require the mandatory criterion of palpable purpura PLUS at least one of four additional criteria: abdominal pain, arthritis/arthralgia, renal involvement (haematuria/proteinuria), or IgA deposits on biopsy. The biopsy finding of IgA deposits satisfies the fourth additional criterion (IgA biopsy). In this case, the child also has two other additional criteria (renal involvement from haematuria, and arthralgia from ankle pain), so she meets criteria without the biopsy. However, the question specifically asks which criterion the biopsy satisfies — the IgA biopsy criterion.
Diagnosis and Investigation
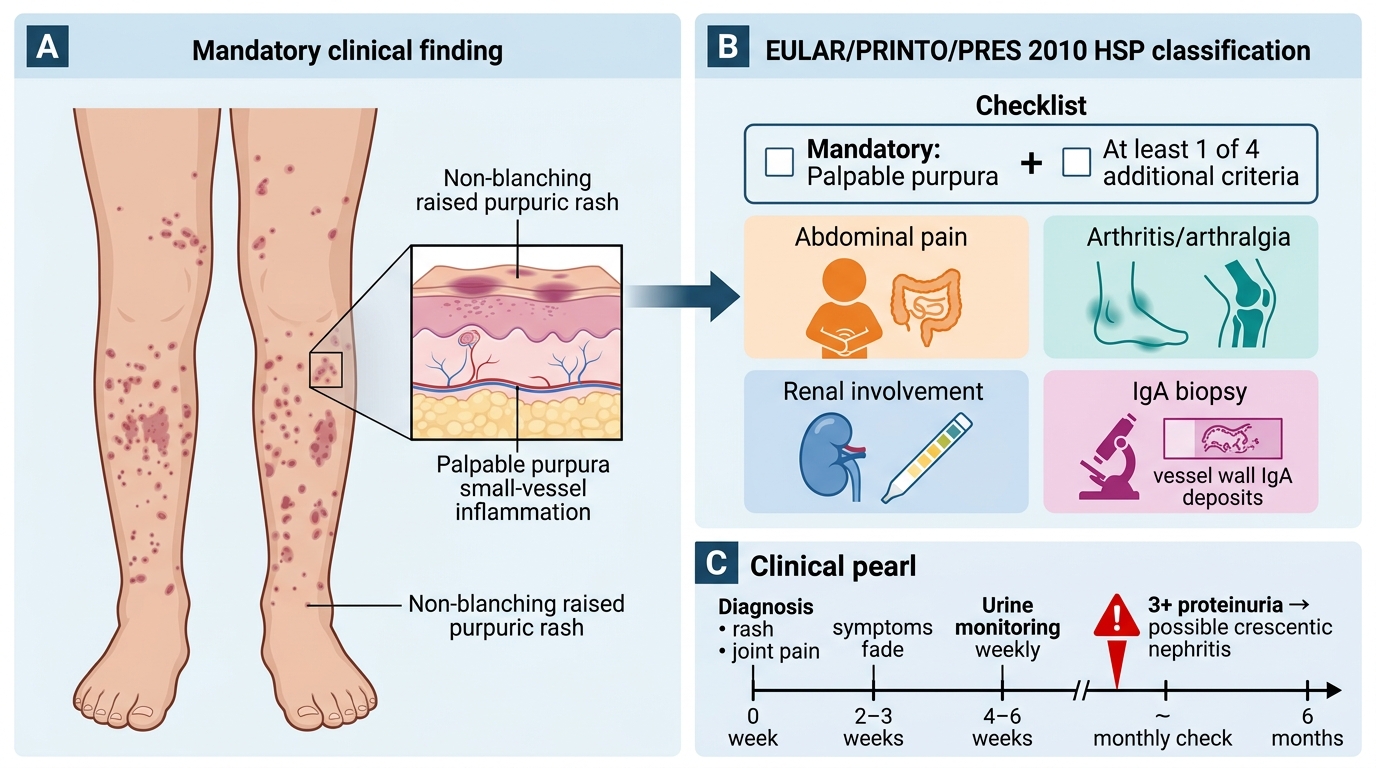
The diagnosis of HSP is primarily clinical, based on the EULAR/PRINTO/PRES 2010 classification criteria, which were developed and validated in a large paediatric multicentre cohort and have replaced the older Michel criteria. These criteria were designed to be applied at the bedside: a child with the right combination of clinical features can be diagnosed and managed without a biopsy in the majority of cases. The central insight is that the criteria are hierarchical — one criterion is mandatory and non-negotiable (the palpable purpura), and at least one from a further set of four is required. When the mandatory criterion is absent or atypical, or when another vasculitis is in the differential, skin or renal biopsy with direct immunofluorescence becomes essential. Understanding this two-tier structure prevents both over-investigation in classic cases and under-investigation in atypical presentations. The criteria require: (1) the mandatory criterion of palpable purpura (lower limb predominant, non-thrombocytopenic), PLUS (2) at least one of four additional criteria.
The four additional criteria are:
• Abdominal pain (diffuse colicky, acute onset)
• Arthritis (any joint) or arthralgia
• Renal involvement (haematuria and/or proteinuria)
• IgA deposits on biopsy (skin or renal)
The diagnosis can be made clinically in the presence of the characteristic rash without biopsy when at least one additional criterion is met. In atypical presentations (e.g., rash on the upper body only, or absent purpura at the time of presentation), skin biopsy with direct immunofluorescence confirming IgA deposits is diagnostic. Skin biopsy is also useful when the diagnosis is uncertain and to exclude other small-vessel vasculitides (e.g., polyarteritis nodosa, ANCA-associated vasculitis).
Investigations to perform:
• Full blood count: platelet count MUST be normal (thrombocytopenia suggests immune thrombocytopenia or another vasculitis; this is a diagnostic exclusion criterion)
• Coagulation screen: normal (excludes disseminated intravascular coagulation in sick patients)
• Urine dipstick + microscopy + protein:creatinine ratio: most important monitoring test — haematuria and proteinuria indicate renal involvement; repeat weekly for 4–6 weeks
• Serum IgA: elevated in ~50% — supportive but not required
• Renal function tests: urea, creatinine, eGFR — particularly if proteinuria is heavy or persistent
• ASOT/anti-DNase B: if streptococcal trigger suspected
• Abdominal ultrasound: if abdominal pain is severe or intussusception is suspected — look for bowel wall thickening, ileo-ileal intussusception, free fluid
• Renal biopsy: indicated for heavy proteinuria (nephrotic range: protein:creatinine >2 or urinary protein >40 mg/m²/hr), declining renal function, or hypertension; reveals IgA mesangial deposits (similar to IgA nephropathy) — graded by ISKDC classification I–VI (class I = minimal change; class II = mesangial proliferation; class IIIa = <50% crescents; class IIIb–V = increasing crescentic glomerulonephritis; class VI = membranoproliferative)
Do NOT order these routinely: ANCA, ANA, anti-dsDNA, complement levels (these are tests for other vasculitides/SLE and are normal in uncomplicated HSP; ordering them unnecessarily leads to confusion).
EULAR/PRINTO/PRES 2010 HSP Classification Criteria
CLINICAL PEARL
The most important lesson in HSP management is this: the skin rash and joint pain tell you the diagnosis, but the urine tells you the prognosis. The purpura may fade and the abdominal pain resolve within 2–3 weeks, leading parents (and clinicians) to believe the illness is over — but renal involvement can appear or worsen up to 6 months after the initial presentation. Every child diagnosed with HSP needs urine dipstick monitoring weekly for 4–6 weeks and then monthly for 6 months. A single missed urine check that contains 3+ proteinuria could mean a child with class IV crescentic nephritis goes undetected until renal failure develops.