Page 5 of 31
PE22.2 | Cyanotic Heart Disease — SDL Guide (Part 2)
Diagnosis and Investigation
A systematic approach to investigation confirms the anatomical diagnosis and guides urgent management decisions. The priority investigations are performed in parallel while PGE1 is administered if duct-dependency is suspected.
1. Hyperoxia test (Nitrogen washout test): place the neonate in 100% FiO₂ for 10–15 minutes. Measure arterial PaO₂ (ABG) or SpO₂. In cardiac cyanosis: PaO₂ remains <150 mmHg (usually <100 mmHg) because right-to-left shunting is anatomical — oxygen cannot access deoxygenated blood. In pulmonary cyanosis (respiratory causes): PaO₂ rises to >200–250 mmHg as ventilation-perfusion mismatch corrects with oxygen. This is the single most useful bedside discriminator of cardiac vs respiratory cyanosis in the neonate.
2. Chest X-ray (CXR): cardiac size, situs, mediastinal width, aortic arch side, and pulmonary vascular markings:
• TOF: boot-shaped heart (coeur en sabot), oligaemic fields
• TGA: egg-on-side/egg-on-a-string, plethoric fields
• TAPVC (supracardiac): figure-of-8 / snowman sign; infracardiac = ground-glass oedema
• Truncus: cardiomegaly, plethoric, right aortic arch (30–35%)
• Tricuspid atresia: left-sided cardiac apex, variable vascularity
3. Electrocardiogram (ECG):
• TOF: right axis deviation (RAD), right ventricular hypertrophy (RVH) — tall R in V1
• TGA: RAD, RVH
• Tricuspid atresia: left axis deviation (LAD), LVH — this is the classic exam discriminator for tricuspid atresia among cyanotic lesions
• TAPVC: RVH, RAD
• Truncus: biventricular hypertrophy
4. Pulse oximetry and four-limb saturations: differential saturations between upper and lower limbs suggest ductal flow; e.g. in coarctation with TGA the lower-body saturation may be higher (ductus supplies upper body with deoxygenated blood in some anatomical variants). Routine SpO₂ screening in neonates (target ≥95% in both upper and lower limbs, ≤3% difference) is recommended for early CHD detection.
5. Echocardiography — the gold standard for anatomical diagnosis. Two-dimensional echo with Doppler defines: cardiac chambers and their size; great vessel origins and connections (ventriculo-arterial concordance/discordance); septal defects (ASD, VSD); ductal patency; pulmonary venous connections; degree of RVOTO; and gradient across stenotic lesions. Neonates with unexplained cyanosis must have urgent echo before definitive management decisions.
6. Arterial blood gas (ABG): metabolic acidosis (raised lactate) indicates cardiac output compromise — a marker of severity. Confirm the partial pressure of oxygen with the hyperoxia test.
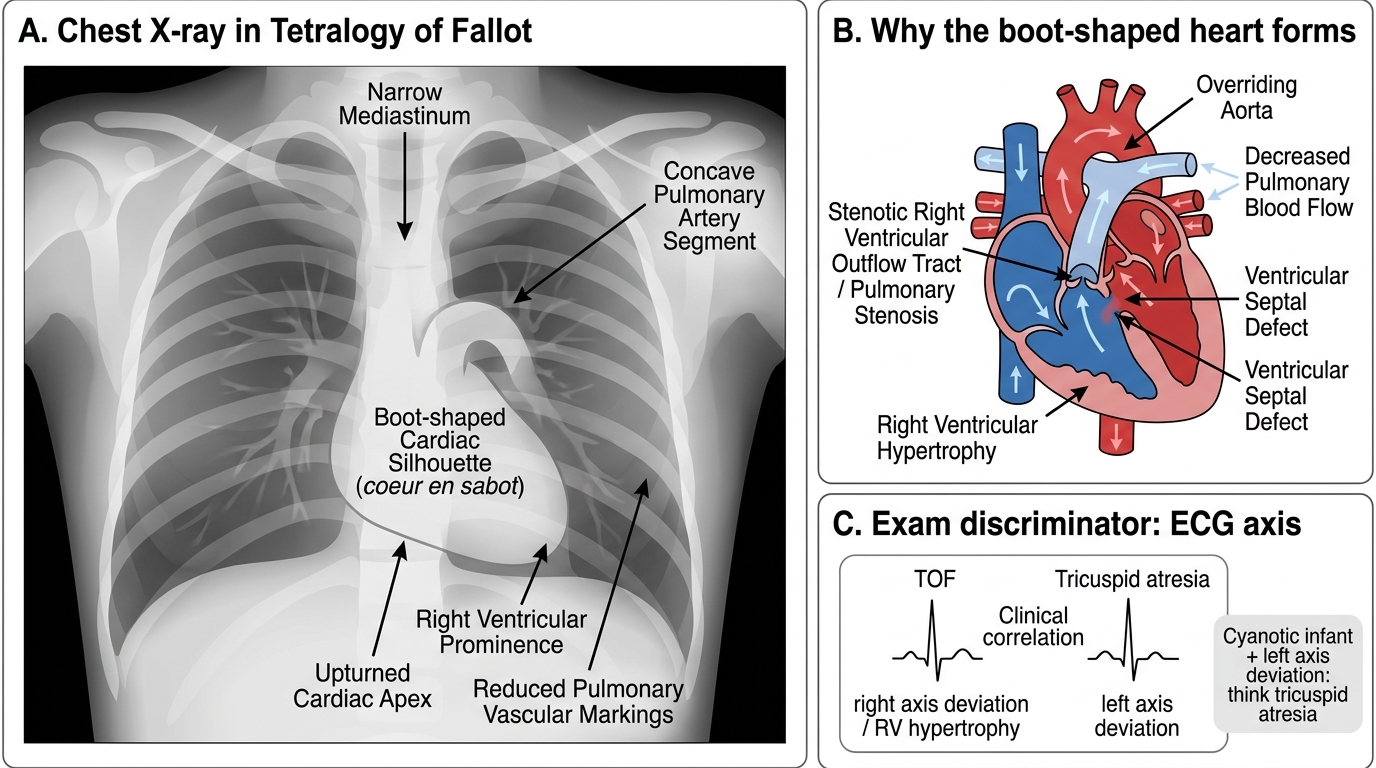
Tetralogy of Fallot: Boot-Shaped Heart on Chest X-Ray
CLINICAL PEARL
Tricuspid atresia is the only cyanotic CHD with LEFT axis deviation on ECG — all others (TOF, TGA, TAPVC) show right axis deviation or biventricular hypertrophy. This is because tricuspid atresia forces all flow through the left ventricle (which hypertrophies markedly), while the right ventricle is hypoplastic. In an exam vignette, if a cyanotic infant has left axis deviation on ECG, think tricuspid atresia first. This is a reliable one-line discriminator that works regardless of the CXR appearance.
Management — Palliation and Definitive Repair
Management of cyanotic CHD is a medical emergency requiring immediate stabilisation, urgent imaging, and timely surgical or catheter-based intervention. The overarching principle is to ensure adequate mixing of oxygenated and deoxygenated blood in mixing lesions, or to restore pulmonary blood flow in obstructive lesions, while preparing for definitive repair. The single most life-saving initial intervention for duct-dependent lesions is prostaglandin E1, which keeps the ductus arteriosus patent and buys hours to days for diagnostic workup and surgical planning. A crucial practical point is that the management sequence must begin immediately, even before echocardiographic confirmation, if the clinical and X-ray picture are consistent with duct-dependent cyanosis — waiting for imaging to confirm before starting PGE1 in a deteriorating neonate is a preventable error. Simultaneously, catheter-based and surgical palliation serve as bridges to definitive repair, which is timed by lesion type and the infant's weight and haemodynamic stability.
Step 1: Immediate stabilisation
- Ensure airway, breathing, circulation.
- Avoid 100% oxygen in mixing lesions (TGA, TAPVC, Truncus): hyperoxia paradoxically increases pulmonary blood flow and worsens pulmonary congestion without increasing systemic saturation.
- Correct metabolic acidosis (sodium bicarbonate if pH <7.1 after ensuring ventilation).
- Correct hypoglycaemia and hypocalcaemia (screen for DiGeorge in truncus/TGA).
Step 2: Prostaglandin E1 (PGE1 / alprostadil) for duct-dependent lesions
PGE1 keeps the ductus arteriosus patent by relaxing ductal smooth muscle via EP receptor stimulation. Dose: 0.05–0.1 mcg/kg/min IV infusion. Start at the lower dose; increase to 0.1 if response is inadequate. Side effects: apnoea (most dangerous — anticipate and have intubation ready), fever, hypotension, jitteriness, hypoglycaemia. Indications include:
• TGA (maintains mixing via ductus, and provides some pulmonary venous return mixing)
• Critical pulmonary stenosis / atresia (ductus is the sole pulmonary blood-flow source)
• Obstructed TAPVC (may buy time by offloading right heart via ductus)
• Hypoplastic left heart syndrome (ductus is the systemic outflow)
• Any neonate with unexplained cyanosis and haemodynamic compromise — start PGE1 empirically while awaiting echo confirmation.
Step 3: Catheter-based palliation
- Rashkind balloon atrial septostomy: enlarges the foramen ovale by balloon catheter, creating an adequate inter-atrial communication. First-line palliation for TGA — by improving mixing between the two parallel circuits, it can raise SpO₂ from 55% to 75–80% in hours. Performed in the catheter lab or at the bedside under echo guidance.
Step 4: Surgical palliation
- Modified Blalock-Taussig (BT) shunt: anastomosis between subclavian artery and pulmonary artery using a Gore-Tex tube graft — increases pulmonary blood flow in decreased-flow lesions (TOF, tricuspid atresia, critical pulmonary stenosis). Used as a bridging procedure in neonates/small infants not yet suitable for complete repair.
- Bidirectional Glenn shunt: SVC-to-pulmonary artery anastomosis — part of the staged Fontan pathway for single-ventricle lesions (tricuspid atresia).
Step 5: Definitive surgical repair (timing is lesion-specific)
| Lesion | Procedure | Timing |
|---|---|---|
| TOF | Total intracardiac repair (VSD patch + RVOTO relief) | 3–6 months of age |
| TGA | Arterial switch operation (Jatene) — great arteries transected and re-anastomosed to correct ventricles + coronary transfer | Within first 2 weeks of life (before LV mass regresses) |
| TAPVC | Re-anastomosis of common pulmonary venous confluence to left atrium | Urgent / emergency |
| Truncus | VSD closure + RV-to-PA conduit | Neonatal period |
| Tricuspid atresia | Staged Fontan (Glenn → total cavopulmonary connection) | Staged over 2–4 years |
Medical management between palliation and repair: propranolol (oral, 1–2 mg/kg/day in 3 divided doses) reduces tet-spell frequency in TOF by relaxing RVOTO infundibular muscle and reducing heart rate. Diuretics if heart failure. Iron supplementation if anaemia (even mild anaemia worsens cyanosis and increases stroke risk).
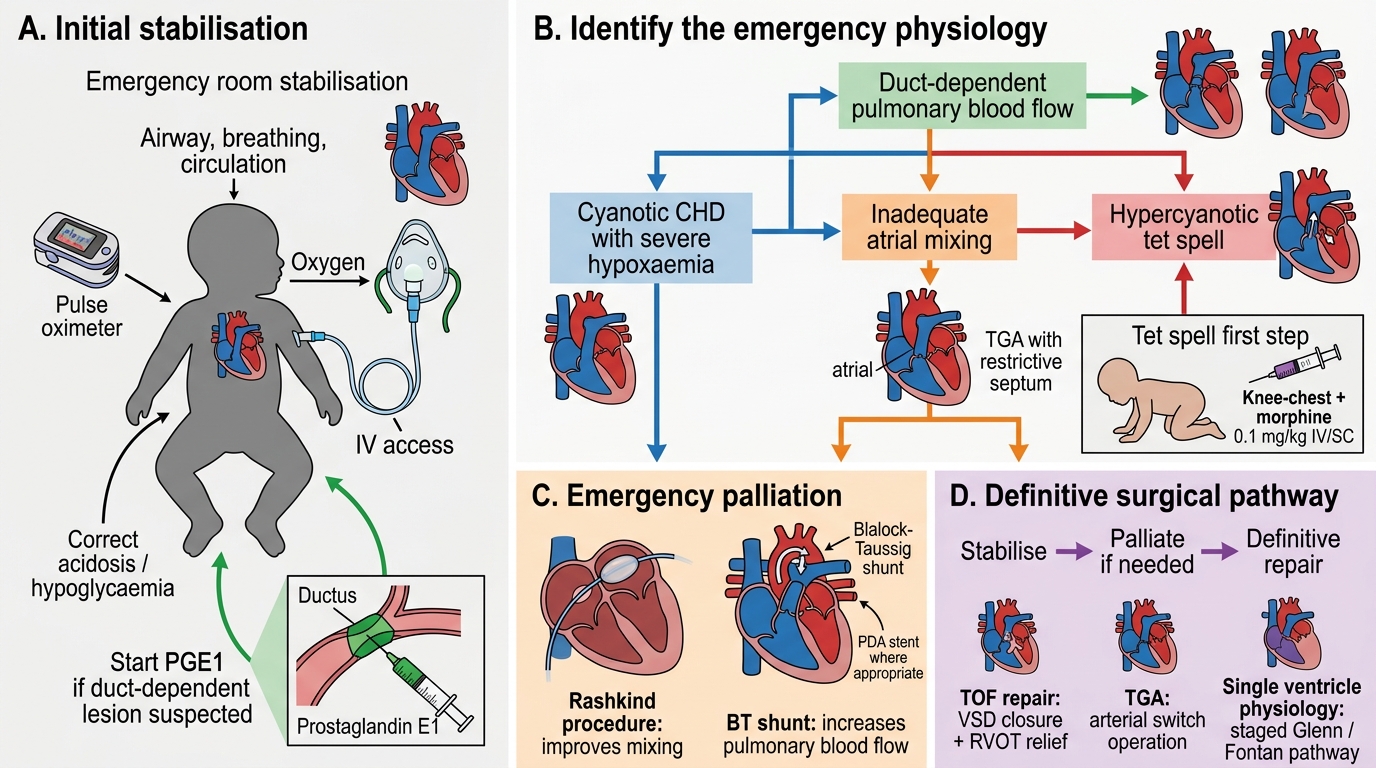
Management Algorithm for Cyanotic Congenital Heart Disease
SELF-CHECK
A 3-month-old infant with known TOF is brought to the emergency room crying inconsolably. He is deeply cyanosed with SpO2 of 55% and is hyperventilating. This is his second such episode. What is the FIRST and most immediately effective management step?
A. Administer 100% oxygen by face mask and observe
B. Place the infant in knee-chest position and administer morphine 0.1 mg/kg IV
C. Start IV propranolol 0.1 mg/kg
D. Prepare for emergency total repair in the operating room
Reveal Answer
Answer: B. Place the infant in knee-chest position and administer morphine 0.1 mg/kg IV
A tet-spell is managed with the knee-chest position FIRST (increases systemic vascular resistance by kinking the femoral arteries, reducing right-to-left shunt) followed by morphine 0.1 mg/kg IV/SC (reduces hyperpnoea, relieves RVOTO spasm, sedates). IV fluids (10 mL/kg bolus) and propranolol follow if needed. Oxygen alone is inadequate — the shunt is anatomical. Emergency surgical repair is not the acute step; the aim is to terminate the spell and plan elective repair. Propranolol is used for spell prevention (oral), not acute termination as first step.
Complications and Long-Term Outcomes
Untreated or late-presenting cyanotic CHD leads to a spectrum of systemic complications that arise from two interconnected processes: chronic arterial hypoxaemia and the compensatory polycythaemia it drives. Even after successful surgical repair, these children require structured long-term surveillance because both the underlying haemodynamic disturbance and the operative intervention itself carry late sequelae. Understanding these complications is not merely academic — the intern or junior resident who recognises a cyanotic CHD child presenting with fever and focal neurological deficits, and immediately considers brain abscess rather than viral illness, is drawing on exactly this knowledge. The paradoxical physiology — polycythaemia that coexists with iron deficiency; a right-to-left shunt that allows both clots and bacteria to reach the brain directly — requires conceptual clarity that this section establishes.

Provided image
Complications of chronic cyanosis and right-to-left shunting:
- Polycythaemia and hyperviscosity: compensatory erythropoiesis elevates haematocrit to 65–75% or higher. Consequences include headache, dizziness, embolic events, and — paradoxically — iron deficiency (iron is consumed by erythropoiesis; iron-deficient red cells are smaller and stiffer → worse viscosity than a normal haematocrit would suggest; screen with MCV and serum ferritin).
- Brain abscess: the most feared CNS complication in older infants and children (typically >2 years). Venous micro-emboli or bacteria in the systemic venous return normally cleared by the pulmonary capillary bed instead bypass directly to the cerebral circulation via the right-to-left shunt. Staphylococcus aureus and anaerobes are common causative organisms. Presents with fever, headache, focal neurological deficit, or seizures in a known cyanotic CHD child. Diagnosis by CT or MRI brain.
- Paradoxical embolism: air, thrombus, or bacteria in peripheral veins can travel directly to the systemic arterial circulation, causing stroke, mesenteric ischaemia, or limb ischaemia. Take particular care to exclude air bubbles from all IV lines in cyanotic CHD.
- Gout: hyperuricaemia from increased red cell turnover and reduced urate excretion from hyperviscosity.
- Haemostatic disturbances: thrombocytopenia (platelet consumption in polycythaemic sludging), prolonged PT/PTT (von Willebrand factor depletion), paradoxically increases bleeding risk alongside clotting risk.
- Infective endocarditis: all structural cyanotic CHD lesions carry risk — prophylaxis with amoxicillin 50 mg/kg (max 2 g) 30–60 min before dental/invasive procedures per IAP/AHA guidelines.
Post-operative surveillance after definitive repair:
• TOF: risk of residual RVOTO, residual VSD, pulmonary regurgitation (after transannular patch repair → long-term RV dilatation), and arrhythmia (ventricular tachycardia — cause of late sudden death).
• TGA: coronary transfer complications, aortic root dilatation, atrial arrhythmia.
• TAPVC: risk of pulmonary venous stenosis at the anastomosis — re-stenosis is the most feared post-operative complication.
• Fontan circulation (tricuspid atresia): protein-losing enteropathy, plastic bronchitis, progressive liver fibrosis, arrhythmia.