Page 29 of 42
PE23.15 | Chronic Liver Disease — SDL Guide
Learning Objectives
- Describe the clinical features of chronic liver disease (CLD) in children including signs of hepatic insufficiency and portal hypertension
- Enumerate the common causes of CLD in children, stratified by age of presentation
- Explain the pathophysiology of hepatic fibrosis and cirrhosis
- Outline the investigation approach for CLD including disease-specific tests
- Describe the management of common causes of CLD including Wilson disease, autoimmune hepatitis, and biliary atresia
- Identify indications for liver transplant in paediatric CLD
INSTRUCTIONS
Chronic liver disease in children is a clinically challenging spectrum that differs fundamentally from adult CLD — the aetiologies, natural history, and therapeutic windows are all different. Biliary atresia, Wilson disease, and autoimmune hepatitis are among the most important treatable causes, and early diagnosis directly improves outcomes. This module builds on your understanding of fulminant hepatic failure (acute presentation) and prepares you to approach the child with long-standing jaundice, hepatosplenomegaly, or growth failure systematically. The skills of recognising clinical signs of CLD (spider angiomata, palmar erythema, ascites, clubbing) and interpreting the diagnostic panel are directly relevant to clinical postings and examinations.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 18 — Diseases of Liver and Biliary System (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 383 — Chronic Liver Disease (textbook)
- ESPGHAN/NASPGHAN guidelines on paediatric liver disease (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 10-year-old boy is referred for evaluation of 'swollen abdomen' of 6 months' duration. On examination he is thin, with yellow sclerae, multiple spider naevi on his chest, palmar erythema, and a tremor of the outstretched hands. His abdomen reveals a firm, nodular liver palpable 4 cm below the costal margin and a massive spleen. There is shifting dullness. His mother mentions that his elder sister had 'liver problems' too. Serum ceruloplasmin comes back at 9 mg/dL. What is the diagnosis, and what is your management plan?
WHY THIS MATTERS
Chronic liver disease in childhood carries a double burden: it causes immediate morbidity through malnutrition and portal hypertension complications, and it threatens long-term survival through progressive cirrhosis and hepatocellular carcinoma. Unlike adult CLD where alcohol is a dominant cause, paediatric CLD has a rich aetiological diversity — biliary atresia, Wilson disease, autoimmune hepatitis, alpha-1 antitrypsin deficiency — many of which are treatable if identified early. The Kasai portoenterostomy for biliary atresia has a time-sensitive window of efficacy (best outcomes before 60 days of age); Wilson disease is curable with copper chelation if caught before cirrhosis is irreversible; autoimmune hepatitis responds to immunosuppression. Recognising the clinical pattern of CLD and initiating the correct diagnostic workup is a foundational paediatric and internal medicine skill.
RECALL
Before proceeding, activate your prior knowledge:
- Portal circulation (from AN): the portal vein drains the gut and spleen into the hepatic sinusoids; obstruction at any level causes portal hypertension and its complications (varices, splenomegaly, ascites).
- Liver synthetic functions (from PY): albumin, clotting factors II/V/VII/IX/X, and bilirubin conjugation are all hepatic. Chronic failure of these functions explains hypoalbuminaemia, coagulopathy, and jaundice in CLD.
- Wilson disease and FHF (from previous SDL pe16-fulminant-hepatic-failure): Wilson disease can present both as acute FHF and as chronic progressive CLD — ceruloplasmin and urinary copper are the primary screening tests.
- Bilirubin fractions (from PY): cholestatic pattern = predominantly conjugated (direct) hyperbilirubinaemia; hepatocellular pattern = mixed. Recognising the pattern narrows the aetiology.
Clinical Presentation of Chronic Liver Disease
The clinical presentation of chronic liver disease (CLD) in children reflects two overlapping pathological processes that worsen in parallel as the disease progresses: hepatic insufficiency (failure of the liver's metabolic and synthetic functions) and portal hypertension (elevated pressure in the portal venous system from intrahepatic or extrahepatic obstruction). In early CLD the child may appear deceptively well, with only mild jaundice or hepatomegaly; it is the combination of signs across systems that raises suspicion for CLD and should prompt structured investigation. Growth failure and malnutrition are particularly prominent in paediatric CLD — the liver is central to nutrient processing, and cholestasis impairs fat-soluble vitamin absorption (vitamins A, D, E, K), causing specific deficiency states on top of generalised wasting.
Signs of hepatic insufficiency:
• Jaundice — scleral icterus and skin yellowing (conjugated hyperbilirubinaemia in cholestatic CLD; mixed in hepatocellular)
• Spider angiomata — dilated arterioles on the upper chest, face, and arms; >5 is clinically significant; caused by oestrogen excess due to impaired hepatic degradation
• Palmar erythema — erythema of the thenar and hypothenar eminences; same oestrogen-related mechanism
• Finger clubbing — digital clubbing and leukonychia (white nails); from hypoxia and portopulmonary connections
• Gynaecomastia — in adolescent boys; impaired oestrogen clearance
• Fetor hepaticus — musty breath in advanced disease
• Coagulopathy — bruising, petechiae; INR elevated
• Hypoalbuminaemia — peripheral oedema, ascites, muscle wasting
• Hepatic encephalopathy — behavioural change, sleep disturbance (usually less acute than in FHF)
Signs of portal hypertension:
• Splenomegaly — firm, non-tender, often massive; causes hypersplenism (anaemia, thrombocytopenia, leucopenia)
• Ascites — fluid in the peritoneal cavity; detected by shifting dullness and fluid thrill
• Caput medusae — dilated periumbilical collateral veins; a striking sign of portal hypertension
• Haematemesis or malaena — from oesophageal or gastric varices (see portal hypertension SDL)
Growth failure and nutritional signs:
• Failure to thrive, weight-for-height below −2 SD, MUAC reduced
• Rickets (vitamin D deficiency from cholestasis-related fat malabsorption)
• Coagulopathy worsened by vitamin K deficiency
• Night blindness (vitamin A deficiency)
⚑ AI image — pending faculty review (auto-QA score 6/10; best of 3 attempts)
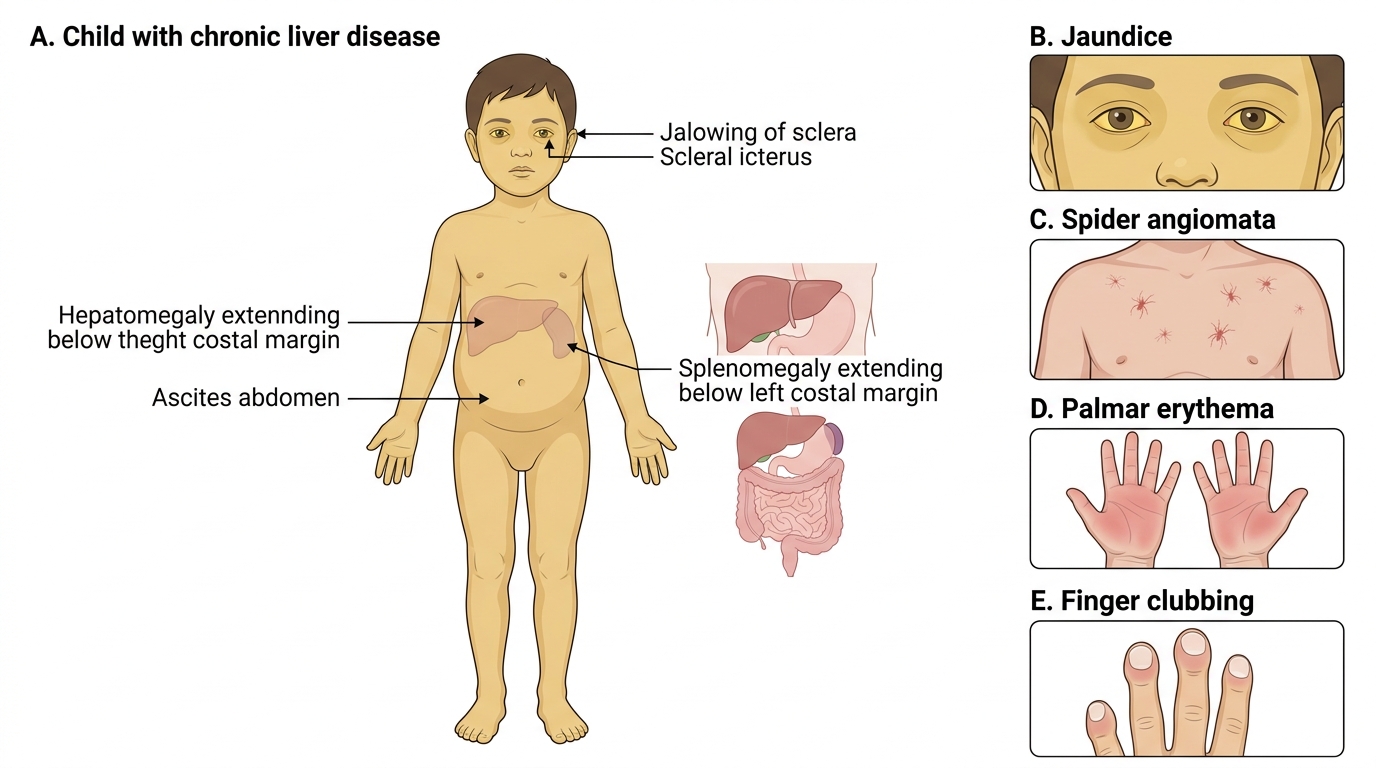
Clinical Signs of Chronic Liver Disease in a Child
Aetiology of CLD in Children
The most powerful first discriminator for aetiology in paediatric CLD is the age at presentation, because different diseases have characteristic onset windows. A neonate with jaundice and acholic stools almost certainly has biliary atresia or another structural biliary problem; a school-age child with tremor, behavioural change, and liver disease has Wilson disease until proven otherwise; an adolescent with elevated IgG and autoantibodies likely has autoimmune hepatitis. Constructing this age-stratified differential before ordering tests prevents both under-investigation (missing treatable causes) and over-investigation (ordering expensive tests unlikely to be positive). Once the age-bracket is established, the investigation panel can be tailored. Genetic and metabolic causes dominate in infancy; immune-mediated and acquired causes dominate in school-age and adolescent years; a subset of causes (Wilson, chronic viral hepatitis, NAFLD) span multiple age groups and require specific screening tests regardless of age.
Neonatal and early infancy (0–3 months):
• Biliary atresia — progressive inflammatory fibro-obliteration of the extrahepatic biliary tree; presents with conjugated hyperbilirubinaemia, acholic (pale/clay-coloured) stools, dark urine; Kasai portoenterostomy is the definitive initial treatment — success rate highest if performed before 60 days of age; liver transplant is eventually needed in most patients
• Neonatal hepatitis / idiopathic neonatal cholestasis — inflammation of hepatocytes causing cholestasis; must rule out metabolic causes
• Alagille syndrome — autosomal dominant (JAG1/NOTCH2 mutations); bile duct paucity on biopsy + 5 features: chronic cholestasis, cardiovascular anomalies (peripheral pulmonic stenosis), butterfly vertebrae, posterior embryotoxon, characteristic facies (broad forehead, deep-set eyes, pointed chin)
Infancy–school age (3 months–8 years):
• Alpha-1 antitrypsin (A1AT) deficiency — PiZZ genotype; misfolded protein accumulates in hepatocytes causing hepatocyte injury (NOT absence of protease inhibitor); liver biopsy shows PAS-diastase-resistant globules in hepatocytes; diagnosis by serum A1AT level + Pi typing
• Glycogen storage diseases (GSD I, III, IV) — hepatomegaly + hypoglycaemia in GSD I; GSD IV can cause progressive fibrosis
• Tyrosinaemia type 1 — fumarylacetoacetate hydrolase deficiency; hepatocellular damage + hepatocellular carcinoma risk; succinylacetone in urine is diagnostic; NTBC (nitisinone) is disease-modifying
School age and adolescence (>5 years):
• Wilson disease — ATP7B gene; copper accumulates in liver, brain, cornea; Kayser-Fleischer rings (on slit-lamp), low ceruloplasmin (<20 mg/dL), elevated 24-hour urinary copper; penicillamine or trientine + zinc
• Autoimmune hepatitis (AIH) — Type 1: ANA + anti-SMA + elevated IgG; Type 2: anti-LKM1 (younger, more severe); responds to prednisolone + azathioprine
• Chronic hepatitis B (HBV) — vertical transmission most common in India; HBsAg positive >6 months = chronic; antiviral therapy (tenofovir, entecavir) for active disease
• Chronic hepatitis C (HCV) — rare in children; transfusion-acquired or vertical; now curable with direct-acting antivirals (DAAs)
• NAFLD/NASH — non-alcoholic fatty liver disease associated with childhood obesity and metabolic syndrome; can progress to cirrhosis
• Primary sclerosing cholangitis (PSC) — often associated with inflammatory bowel disease; MRCP shows multifocal biliary strictures
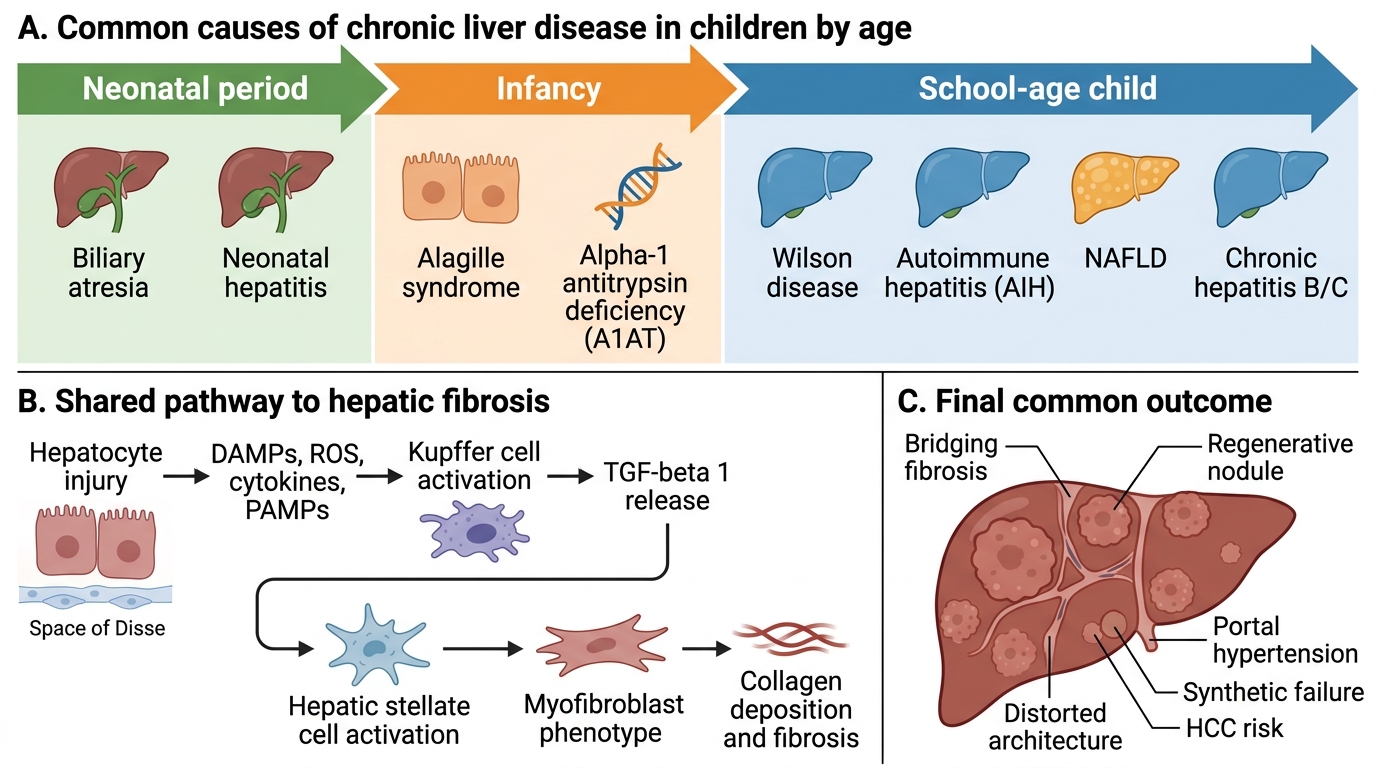
Age-Stratified Causes and Fibrosis Pathway in Pediatric Chronic Liver Disease
Pathophysiology: Fibrosis and Cirrhosis
Regardless of the initial insult — copper accumulation in Wilson disease, misfolded A1AT protein retention in hepatocytes, immune attack in autoimmune hepatitis, or bile acid toxicity in biliary atresia — the final common pathway of CLD is progressive hepatic fibrosis driven by a stereotyped cellular response. Understanding this shared mechanism explains why cirrhosis looks the same histologically regardless of aetiology, and why the complications of advanced CLD (portal hypertension, hepatic synthetic failure, hepatocellular carcinoma) are broadly similar across disease groups. The key effector cell in hepatic fibrogenesis is the hepatic stellate cell (HSC), normally a quiescent fat-storing cell in the perisinusoidal space of Disse. When hepatocytes are injured and die, they release damage signals including reactive oxygen species, cytokines (TNF-alpha, IL-6), and pathogen-associated molecular patterns (PAMPs from gut-derived bacteria). These signals activate HSCs into a myofibroblast-like phenotype.
The fibrogenesis cascade:
1. Hepatocyte injury (any cause) → necroinflammation and release of DAMPs/PAMPs
2. Kupffer cell (hepatic macrophage) activation → secretion of TGF-beta 1 (the principal pro-fibrotic cytokine) and PDGF
3. Hepatic stellate cell (HSC) activation → myofibroblastic transformation, proliferation, contractility
4. Activated HSCs secrete Type I and III collagen, matrix metalloproteinase inhibitors (TIMPs), and further cytokines → extracellular matrix (ECM) accumulation in the space of Disse → sinusoidal fibrosis
5. Progressive collagen deposition → distortion of hepatic architecture → nodular regeneration = cirrhosis
Consequences of cirrhosis:
• Architectural distortion → increased intrahepatic vascular resistance → portal hypertension (oesophageal varices, splenomegaly, ascites, hepatorenal syndrome)
• Reduced hepatocyte mass → impaired synthetic function (hypoalbuminaemia, coagulopathy, hypoglycaemia) and impaired detoxification (hyperammonaemia, drug accumulation)
• Activated HSCs → liver stiffness detectable on elastography (FibroScan) even before cirrhosis is radiologically apparent
• Cirrhosis is largely irreversible — hence the urgency of treating underlying causes before end-stage disease; early aetiology-specific therapy can halt fibrosis progression and in some cases allow partial regression
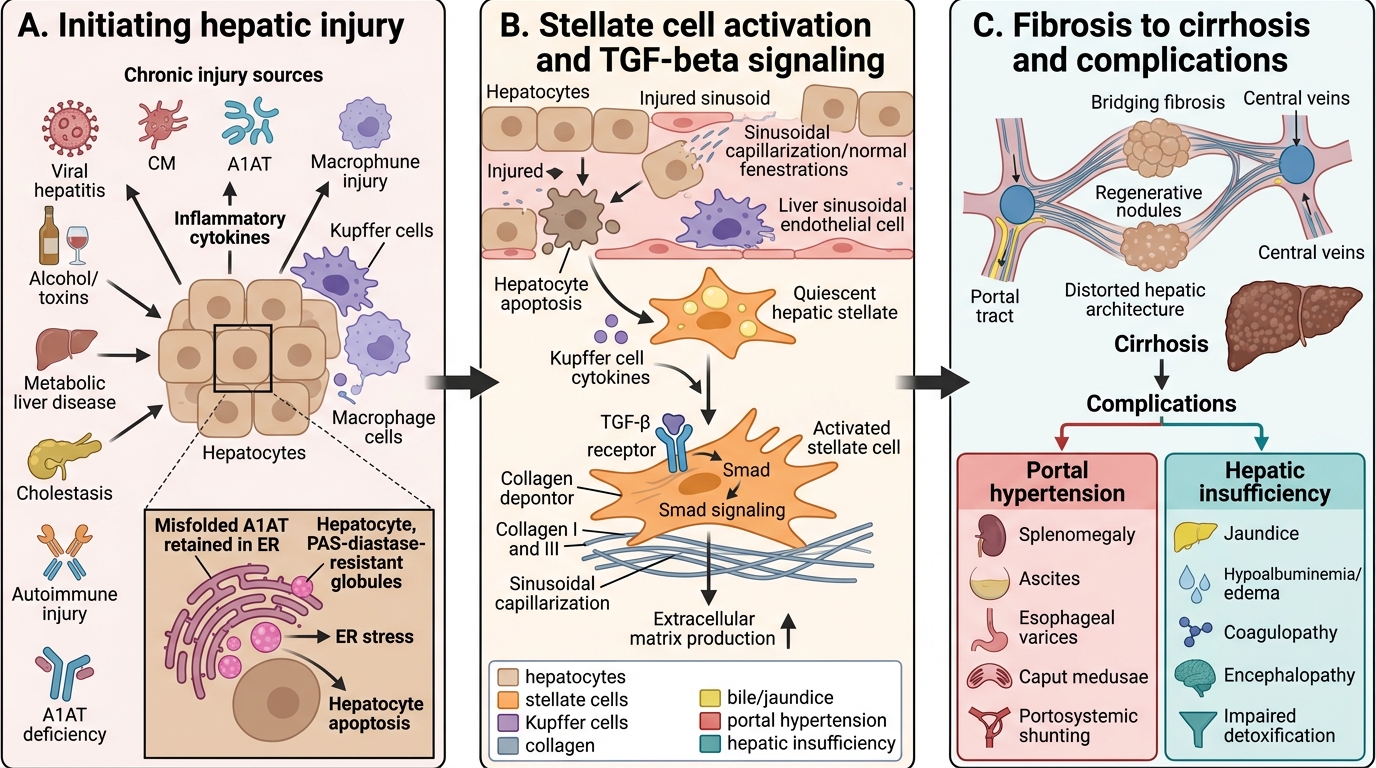
Progression of Chronic Hepatic Injury to Cirrhosis
SELF-CHECK
A 6-year-old girl is found to have chronic liver disease. Liver biopsy shows PAS-diastase-resistant globules within hepatocytes. Serum A1AT level is very low. What is the mechanism of liver injury in this condition?
A. Absence of A1AT protease inhibitor allows unchecked neutrophil elastase activity, destroying hepatocytes
B. Misfolded A1AT protein accumulates within hepatocytes, causing endoplasmic reticulum stress and hepatocyte apoptosis
C. A1AT activates hepatic stellate cells directly, bypassing inflammation
D. A1AT deficiency causes bile acid accumulation leading to cholestatic liver disease
Reveal Answer
Answer: B. Misfolded A1AT protein accumulates within hepatocytes, causing endoplasmic reticulum stress and hepatocyte apoptosis
A1AT deficiency causes hepatic injury through misfolded protein accumulation in hepatocytes, not through absence of protease inhibitory function (which causes the lung disease). The misfolded PiZ protein is retained in the ER of hepatocytes, causing ER stress, hepatocyte apoptosis, and progressive fibrosis. The PAS-diastase-resistant globules on biopsy represent this retained protein. Lung disease in A1AT deficiency is from absent protease protection, but liver disease is a distinct, protein-misfolding mechanism.
CLINICAL PEARL
The Kasai window — and why neonatal jaundice must never be dismissed as 'physiological' beyond 14 days: In biliary atresia, the Kasai portoenterostomy (hepatoportoenterostomy) achieves bile drainage in 80–90% of cases if performed before 60 days of age, dropping to <30% after 90 days. The operation anastomoses the jejunum directly to the hepatic hilum, bypassing the obliterated extrahepatic ducts. Any conjugated jaundice persisting beyond 14 days of life must be evaluated urgently — a direct bilirubin >20% of total bilirubin is the threshold for cholestasis workup. Pale (acholic) stools and dark urine in a jaundiced neonate make biliary atresia the working diagnosis until proven otherwise. Missing this window condemns the child to early cirrhosis and transplant at a younger age.