Page 30 of 42
PE23.15 | Chronic Liver Disease — SDL Guide (Part 2)
Diagnosis and Investigation
Investigation of paediatric CLD is organised in two tiers: a first-tier screen applied to all children to confirm CLD and establish severity, and a second-tier aetiology-specific panel guided by clinical findings and age. The pattern of LFT abnormalities provides the first interpretive clue — a cholestatic pattern (predominantly elevated bilirubin and alkaline phosphatase, GGT) suggests biliary disease (atresia, Alagille, PSC, choledochal cyst), while a hepatocellular pattern (predominantly elevated ALT/AST, with relatively modest bilirubin) suggests parenchymal disease (Wilson, AIH, viral hepatitis, NAFLD). Neither pattern is exclusive, and mixed patterns are common in advanced disease where all functions are compromised. The gold standard for grading fibrosis and identifying aetiology-specific histological features (bile duct paucity in Alagille, interface hepatitis in AIH, PAS-diastase globules in A1AT) remains liver biopsy, although non-invasive fibrosis markers (elastography, APRI score) are increasingly used to avoid biopsy in low-risk situations.
First-tier investigations (all patients with suspected CLD):
• Liver function tests: Bilirubin (total + direct), ALT, AST, GGT, alkaline phosphatase, albumin, total protein
• Coagulation profile: PT/INR, APTT (synthetic function; cannot be corrected by Vitamin K alone in true hepatic failure)
• Complete blood count: Anaemia (chronic disease/haemolysis), thrombocytopenia (hypersplenism), leucopenia
• Renal function: Creatinine, electrolytes
• Blood glucose: Hypoglycaemia in severe disease
Aetiology-specific investigations:
| Disease | Key investigation | Diagnostic finding |
|---|---|---|
| Wilson disease | Serum ceruloplasmin; 24-h urine copper; slit-lamp | Ceruloplasmin <20 mg/dL; urinary copper >100 µg/24h; KF rings |
| Autoimmune hepatitis | ANA, anti-SMA (Type 1); anti-LKM1 (Type 2); serum IgG | Positive autoantibodies + elevated IgG |
| Biliary atresia | USG (absent gall bladder or triangular cord sign); HIDA scan; intraoperative cholangiogram | Absent bile flow on HIDA + cholangiography |
| Alpha-1 antitrypsin deficiency | Serum A1AT level; Pi typing | PiZZ genotype + low A1AT (<15% normal) |
| Chronic HBV | HBsAg, HBeAg, anti-HBc IgG, HBV DNA | HBsAg positive >6 months |
| Chronic HCV | Anti-HCV antibody; HCV RNA | Positive RNA >6 months |
| NAFLD | USG (echogenic liver); ALT; liver biopsy for NASH | Steatosis on USG; NASH = steatosis + inflammation + fibrosis |
| Alagille syndrome | JAG1/NOTCH2 gene sequencing; biopsy (bile duct paucity) | <0.9 bile ducts per portal tract |
Imaging:
• Ultrasound abdomen: Liver size/echogenicity, spleen size, ascites, portal vein diameter and flow, hepatic vascularity; can detect cirrhosis (irregular surface, nodular parenchyma) and biliary dilatation
• MRCP (Magnetic Resonance Cholangiopancreatography): Biliary tree evaluation in PSC, choledochal cyst
• Liver elastography (FibroScan): Non-invasive fibrosis staging by measuring liver stiffness (kPa); F0–F4 stages
Liver biopsy:
• Still the gold standard for definitive aetiology (histology) and fibrosis grading (METAVIR F0–F4 or Ishak scoring)
• Performed percutaneously under USG guidance; risks: bleeding (correct INR >1.5 before biopsy), bile leak
SELF-CHECK
A 3-year-old child is referred for evaluation of persistent conjugated hyperbilirubinaemia since the neonatal period with pale stools. She has now developed hepatosplenomegaly and ascites. Eye examination shows posterior embryotoxon. Genetic testing reveals a JAG1 mutation. Which investigation finding is MOST consistent with this diagnosis?
A. Serum ceruloplasmin <20 mg/dL and elevated 24-h urinary copper
B. Liver biopsy showing <0.9 bile ducts per portal tract (bile duct paucity)
C. Anti-smooth muscle antibody (anti-SMA) positive with elevated IgG
D. PAS-diastase-resistant globules in hepatocytes on liver biopsy
Reveal Answer
Answer: B. Liver biopsy showing <0.9 bile ducts per portal tract (bile duct paucity)
The clinical picture — conjugated jaundice since neonatal period, posterior embryotoxon on eye examination, JAG1 mutation — is diagnostic of Alagille syndrome. The histological hallmark is bile duct paucity on liver biopsy: <0.9 interlobular bile ducts per portal tract. Low ceruloplasmin/urinary copper is Wilson disease. Anti-SMA + elevated IgG is autoimmune hepatitis. PAS-diastase globules is A1AT deficiency. Alagille is a NOTCH signalling defect causing failure of intrahepatic bile duct development.
Management of CLD
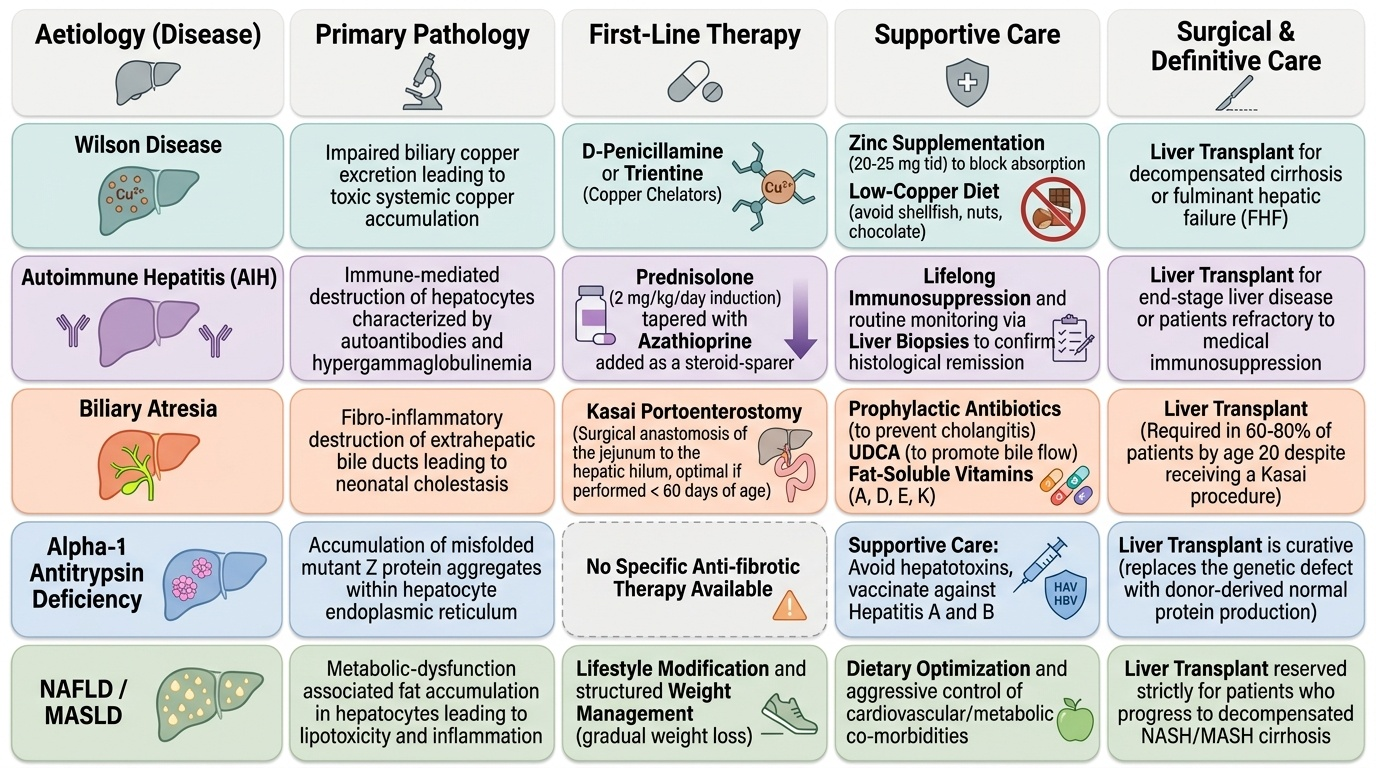
The management of paediatric CLD is built on three pillars: (1) aetiology-specific therapy to halt or reverse the underlying disease process before cirrhosis becomes irreversible; (2) nutritional and complication management to sustain growth and prevent morbidity from portal hypertension, coagulopathy, and fat-soluble vitamin deficiency; and (3) liver transplant as the definitive therapy for end-stage disease or for conditions where no medical therapy is effective. The order of these interventions matters: aetiology-specific therapy is the highest priority in non-cirrhotic or early-cirrhotic disease; once cirrhosis is established and decompensated, transplant evaluation takes precedence. Nutritional optimisation must begin from the point of diagnosis and continue throughout the disease course, because malnutrition worsens outcomes from both the underlying liver disease and from transplant surgery.
Provided image
Aetiology-specific treatments:
- Wilson disease: First-line = penicillamine (D-penicillamine, a copper chelator; adverse effects include nephrotoxicity, bone marrow suppression, lupus-like syndrome — requires monitoring) OR trientine (better tolerated, preferred in many centres). Zinc (20–25 mg elemental zinc three times daily) blocks intestinal copper absorption and is used for maintenance and in mild/asymptomatic disease. Diet: avoid copper-rich foods (shellfish, nuts, chocolate, liver). Liver transplant for decompensated cirrhosis or Wilson FHF not responding to chelation.
- Autoimmune hepatitis (AIH): Prednisolone (2 mg/kg/day, max 40 mg/day) as induction, with azathioprine added after initial response (as steroid-sparer); most children require lifelong immunosuppression. Liver biopsy confirming response is recommended. Relapse on dose reduction requires re-titration.
- Biliary atresia: Kasai portoenterostomy — surgical anastomosis of jejunum to hepatic hilum; best before 60 days of age. Post-Kasai: prophylactic antibiotics (prevent ascending cholangitis), ursodeoxycholic acid (UDCA, to enhance bile flow), fat-soluble vitamins (A, D, E, K) supplementation. Liver transplant is required in 60–80% of biliary atresia patients by age 20 despite Kasai.
- Alpha-1 antitrypsin deficiency: No specific anti-fibrotic therapy; management is supportive (avoid hepatotoxins, vaccinate against hepatitis A and B). Liver transplant is curative (the transplanted liver has normal A1AT gene → normalises serum A1AT).
- Chronic HBV: Tenofovir or entecavir (in children ≥2 years with active viral replication and elevated ALT or significant fibrosis); interferon-alpha in selected patients. Treatment goal: HBeAg seroconversion and viral suppression.
- Chronic HCV: Now curable with direct-acting antivirals (DAAs) — sofosbuvir-based regimens (approved from age ≥3 years); >95% sustained virological response (SVR = cure).
- NAFLD: Lifestyle modification (dietary change + physical activity for weight reduction); no approved pharmacotherapy in children. Vitamin E used in NASH with significant fibrosis in some guidelines.
Nutritional management (all causes of CLD):
• High-calorie diet (130–150% of normal daily requirement in severe CLD)
• Supplementation: fat-soluble vitamins A, D, E, K (especially in cholestatic CLD)
• Nasogastric tube feeding if oral intake inadequate
• Sodium restriction and diuretics (spironolactone ± furosemide) for ascites
• Lactulose for HE episodes
Liver transplantation:
• Indications: decompensated cirrhosis (refractory ascites, encephalopathy, bleeding), failed Kasai (biliary atresia), Wilson FHF, A1AT deficiency with cirrhosis, fulminant AIH not responding to steroids, hepatocellular carcinoma
• PELD score (Pediatric End-Stage Liver Disease) used for prioritisation in children <12 years; MELD for older children/adolescents
• Living related donor transplant is common in India (parental donation)
Monitoring for hepatocellular carcinoma (HCC):
• 6-monthly USG + serum AFP in children with cirrhosis from HBV, tyrosinaemia, or metabolic CLD
SELF-CHECK
A 9-year-old boy with Wilson disease and compensated cirrhosis is started on penicillamine. Six months later he develops worsening neurological symptoms with new tremors. What is the MOST likely explanation for this deterioration?
A. Penicillamine has failed to chelate copper and the disease is progressing naturally
B. Paradoxical neurological worsening from initial copper mobilisation by penicillamine — a recognised side effect in neurologically active Wilson disease
C. Penicillamine nephrotoxicity causing uraemic encephalopathy
D. The child has developed autoimmune hepatitis superimposed on Wilson disease from penicillamine-induced lupus
Reveal Answer
Answer: B. Paradoxical neurological worsening from initial copper mobilisation by penicillamine — a recognised side effect in neurologically active Wilson disease
Paradoxical neurological deterioration after starting penicillamine is a well-recognised phenomenon in Wilson disease patients with neurological involvement — the initial burst of copper mobilisation from tissues can transiently worsen neurological symptoms before improvement occurs. This is why trientine (or zinc as primary therapy) is preferred in neurologically active Wilson disease, as it mobilises copper more gradually. The deterioration is not penicillamine nephrotoxicity (which causes proteinuria, not neurological symptoms) or autoimmune hepatitis (which would not present as new tremors). Recognising this paradox prevents premature discontinuation of an effective therapy.
Self-Assessment: Chronic Liver Disease
Test your understanding with these age-stratified clinical scenarios.
Scenario 1 (Neonate): A 6-week-old infant is referred for persistent jaundice since birth. Stools are pale and urine is dark. Total bilirubin is 9.8 mg/dL with direct bilirubin 7.2 mg/dL. Ultrasound shows absence of the gall bladder fossa and a triangular cord sign. What is the diagnosis, what is the urgency, and what is the treatment?
Key answer points: Biliary atresia. This is an emergency — Kasai portoenterostomy must be performed as soon as possible, ideally before 60 days of age. Delay beyond 90 days markedly reduces operative success rates. Refer immediately to a paediatric surgical centre with hepatobiliary expertise. If successful, continue UDCA, fat-soluble vitamin supplementation, and prophylactic antibiotics post-Kasai.
Scenario 2 (School age): A 12-year-old girl presents with fatigue, jaundice, and elevated ALT (320 U/L). She has no history of blood transfusion or family history of liver disease. ANA is positive (1:160), anti-SMA is positive, and serum IgG is 3.8 g/dL (normal <1.8 g/dL). Liver biopsy shows interface hepatitis with plasma cell infiltration. What is the diagnosis, and how do you manage her?
Key answer points: Autoimmune hepatitis Type 1 (ANA + anti-SMA + elevated IgG). Treatment: prednisolone 2 mg/kg/day (max 40 mg) as induction; once ALT normalises, add azathioprine and taper prednisolone. Most children require lifelong immunosuppression. Regular monitoring of LFTs, CBC (azathioprine myelosuppression), and bone density (steroid osteoporosis).
Scenario 3 (Adolescent): A 15-year-old boy with known chronic hepatitis B (HBsAg positive for 3 years, acquired vertically) has HBV DNA of 2 × 10⁶ IU/mL, ALT three times the upper limit of normal, and liver biopsy showing moderate fibrosis (F2/F4). Should he be treated, and with what?
Key answer points: Yes — active viral replication (HBV DNA detectable) + elevated ALT + significant fibrosis (F2) = indication for antiviral therapy in NMC/ESPGHAN guidelines. First-line: tenofovir disoproxil fumarate (≥12 years) or entecavir (≥2 years) for children. Goal: suppress HBV DNA to undetectable, normalise ALT, and halt fibrosis progression. Treat duration is typically long-term (indefinite for cirrhotic patients; ≥1 year after HBeAg seroconversion in non-cirrhotic HBeAg-positive patients).