Page 16 of 34
PE26.7 | Hemophilia — SDL Guide
Learning Objectives
- Describe the clinical features of hemophilia A and B in children, distinguishing haemarthrosis and deep muscle bleeds from platelet-type bleeding
- Explain the aetiology and classification: factor VIII deficiency (hemophilia A) and factor IX deficiency (hemophilia B), both X-linked recessive, and their impact on the intrinsic coagulation pathway
- Interpret the characteristic coagulation profile — prolonged aPTT with normal PT, platelet count, and bleeding time — and identify confirmatory factor assays
- Apply weight-based factor replacement dosing formulae and outline the principles of management including prophylaxis, DDAVP use in mild hemophilia A, and avoidance of intramuscular injections and aspirin
INSTRUCTIONS
Hemophilia is the prototypic disorder of secondary haemostasis — understanding it illuminates the entire coagulation cascade, explains why a patient bleeds into joints rather than skin, and provides a framework for reasoning about all hereditary coagulopathies. The dosing formulae, genetic counselling implications, and clinical management principles taught here are directly examinable and directly applicable in paediatric ward and emergency settings.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 16 (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 505 (textbook)
- World Federation of Hemophilia (WFH) Guidelines for the Management of Hemophilia, 3rd ed., 2020 (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
An 8-year-old boy is brought to the paediatric OPD by his mother because his left knee has been painful and swollen for two days following a minor fall in the school playground. She mentions that he has had similar episodes three times before — always after minor injuries — and that his maternal uncle has a 'blood problem'. On examination, the knee is warm, swollen, and tender with limited flexion; no petechiae or purpura are visible anywhere. His CBC shows Hb 11.2 g/dL, platelets 220,000/µL, and a normal white count. The coagulation screen reveals aPTT 76 seconds (normal <35 s), with a completely normal prothrombin time (PT). The bleeding time is normal. Why is this boy's aPTT the only abnormal test — and what is draining blood into his joint?
WHY THIS MATTERS
Hemophilia is the most important hereditary coagulation disorder a paediatrician encounters. Recognising the characteristic deep-tissue bleeding phenotype (haemarthrosis, muscle haematomas) and interpreting the isolated prolonged aPTT correctly are core clinical competencies. Errors in diagnosis — missing hemophilia in a child presenting with 'joint pain', or administering an intramuscular injection (which can cause a life-threatening muscle haematoma in a haemophiliac) — have serious consequences. Understanding the X-linked recessive genetics is equally important for genetic counselling of the family. This module also teaches the factor replacement dosing formulae, which are directly examinable and clinically essential.
RECALL
Before proceeding, recall these essentials from your preclinical physiology:
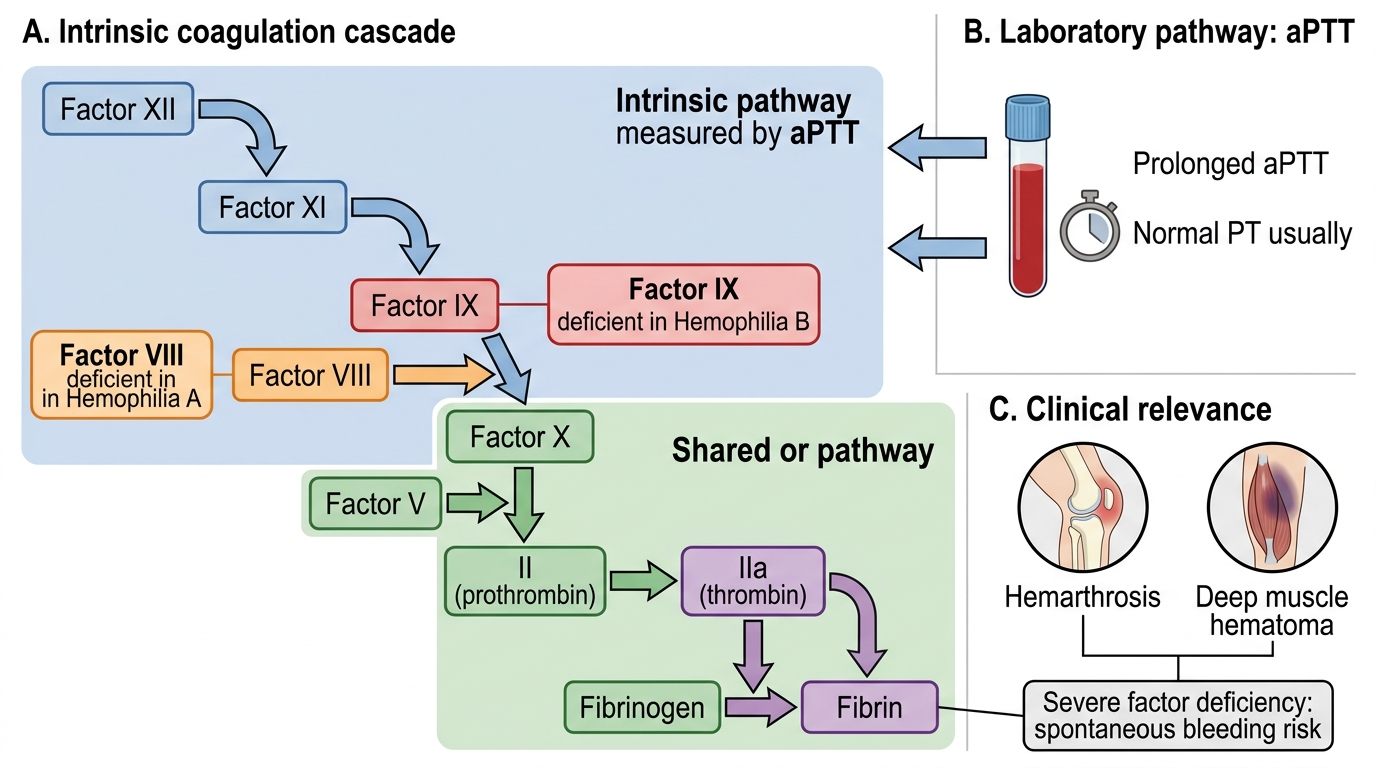
- Coagulation cascade: Two convergent pathways — intrinsic (contact activation) pathway (factors XII, XI, IX, VIII) and extrinsic (tissue factor) pathway (factor VII) — both activating factor X in the common pathway (X, V, II, I).
- aPTT vs PT: aPTT tests the intrinsic + common pathway; PT tests the extrinsic + common pathway. A prolonged aPTT with normal PT indicates a defect in the intrinsic pathway only (factors XII, XI, IX, or VIII).
- Primary vs secondary haemostasis: Primary = platelet plug (tested by platelet count and bleeding time — both normal in hemophilia); Secondary = fibrin clot formation (tested by aPTT and PT — aPTT prolonged in hemophilia).
- X-linked recessive: Gene on the X chromosome; males (XY) with one defective copy are affected; females (XX) with one defective copy are carriers but usually asymptomatic because the other X provides sufficient factor.
Clinical Presentation of Hemophilia
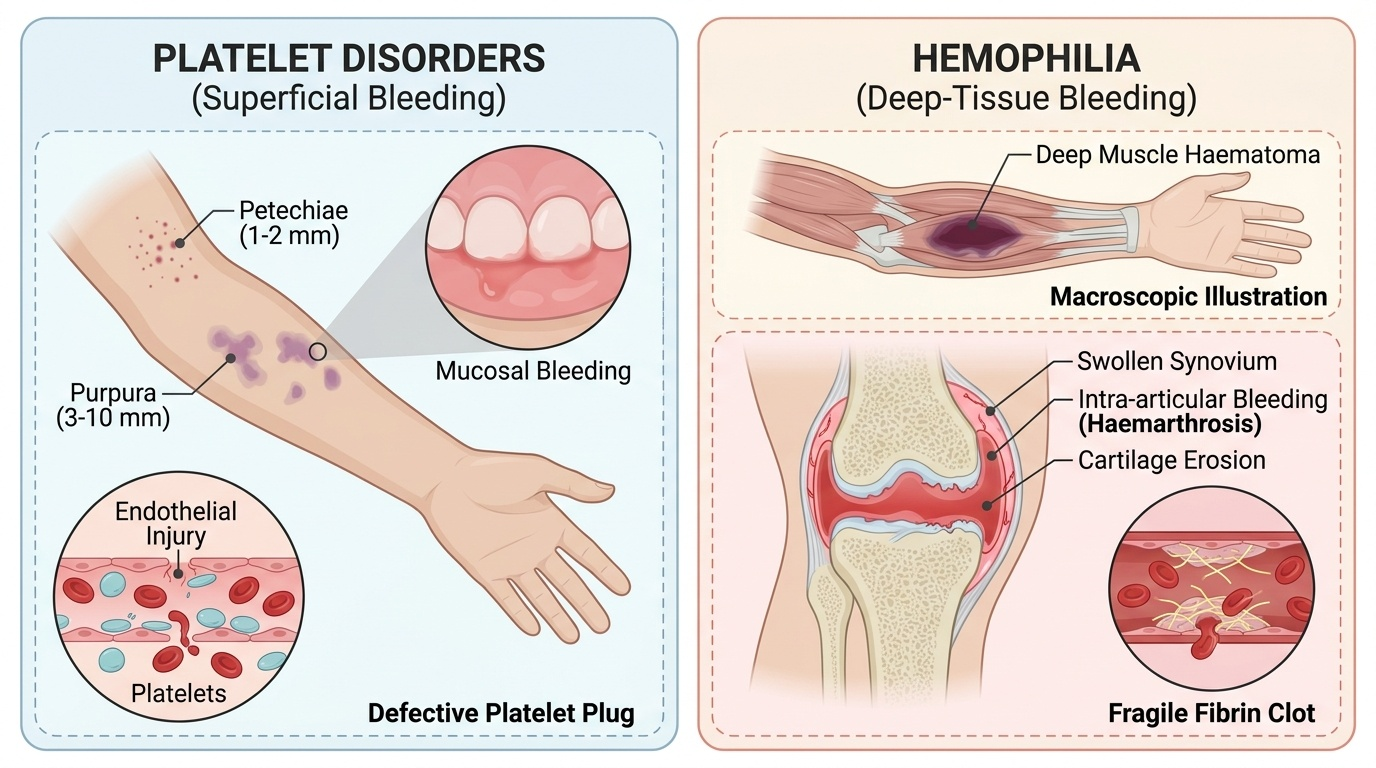
The clinical presentation of hemophilia is dominated by deep-tissue bleeding — a hallmark that immediately distinguishes it from platelet disorders, which produce superficial skin and mucosal bleeding (petechiae, purpura). The prototypical bleeds of hemophilia are haemarthrosis (bleeding into joint spaces) and deep muscle haematomas, which together occur because haemostasis at sites of deeper vascular injury depends entirely on the fibrin clot rather than the platelet plug. Without adequate factor VIII or IX, the intrinsic pathway cannot generate sufficient thrombin, and the clot that forms is fragile and insufficient to contain bleeding into high-pressure spaces like joints.
Provided image
Haemarthrosis is the most characteristic and functionally disabling feature of severe hemophilia. The knee is the most commonly affected joint, followed by the ankle and elbow. A haemarthrotic joint presents as warm, painful, and swollen, with a characteristic 'target joint' phenomenon — repeated bleeds into the same joint lead to chronic synovitis, progressive cartilage destruction, and eventually haemophilic arthropathy with fixed deformity and contracture. This long-term disability is the principal reason prophylactic factor replacement is now preferred over on-demand therapy for severe hemophilia.
The severity of hemophilia determines the bleeding pattern:
• Severe (<1% factor activity): Spontaneous bleeds without identifiable trauma, including into joints, muscles, and the central nervous system; the most devastating complications are intracranial haemorrhage and iliopsoas haematoma (which can compress the femoral nerve).
• Moderate (1–5% factor activity): Bleeds typically require minor trauma to precipitate, though some spontaneous joint bleeds occur.
• Mild (5–40% factor activity): Bleeds occur only with significant trauma or surgery; patients may go undiagnosed until an operative procedure reveals unexpected haemorrhage.
In the neonatal period, hemophilia may present as scalp cephalhaematoma after delivery, prolonged bleeding from the umbilical stump, or unexpected intracranial haemorrhage. Circumcision in an undiagnosed haemophiliac is a classic scenario prompting diagnosis. An important clinical rule: intramuscular (IM) injections are contraindicated in known or suspected hemophilia — the muscle cannot contain the haematoma, which can expand causing compartment syndrome or compressive neuropathy. All vaccines should be given subcutaneously (SC) with firm pressure at the site for ≥5 minutes.
Aetiology, Classification and Pathophysiology
Hemophilia results from mutations in the genes encoding two critical intrinsic pathway coagulation factors, both located on the X chromosome, making both forms X-linked recessive. Hemophilia A, caused by mutations in the gene encoding factor VIII (FVIII), is the most common hereditary coagulopathy — affecting approximately 1 in 5,000 male births. Hemophilia B (also called Christmas disease), caused by mutations in the gene encoding factor IX (FIX), is less common, affecting approximately 1 in 30,000 male births. A third and much rarer form, Hemophilia C (factor XI deficiency), is autosomal recessive and generally milder — it will not be the focus of this module.
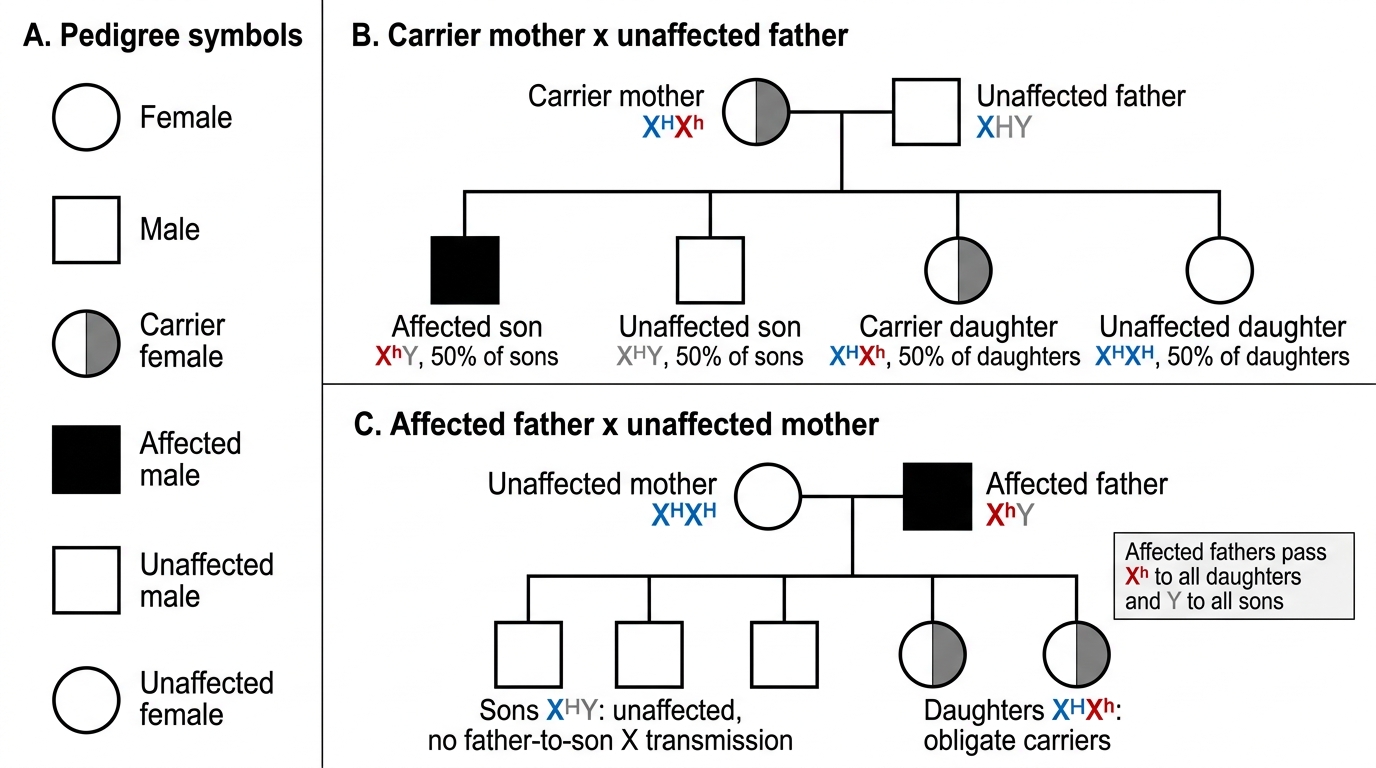
Because both genes reside on the X chromosome (at Xq28 for factor VIII and Xq27 for factor IX), the inheritance pattern is X-linked recessive:
• Carrier females (heterozygous, X^n X) carry one normal and one defective allele; they have ~50% of normal factor activity and are typically asymptomatic.
• Affected males (hemizygous, X^n Y) have only one X chromosome — if it carries the defective allele, no functional factor is produced.
• A carrier mother transmits the defective X to 50% of sons (who are affected) and 50% of daughters (who become carriers).
• Approximately 30% of hemophilia cases arise from de novo mutations — meaning a mother with no family history can have an affected son, which is critical for counselling.
The pathophysiological mechanism of hemophilia lies within the intrinsic pathway, specifically the tenase complex: factor IXa (activated by factor XIa) assembles on phospholipid membranes with factor VIIIa as a cofactor to activate factor X. Without adequate factor VIII or IX, this tenase complex is non-functional, factor X activation is severely impaired, and thrombin generation is dramatically reduced. The result is a clot that cannot withstand mechanical stress — it forms initially (because the extrinsic pathway is intact) but is fragile, thin, and easily disrupted by the mechanical forces in joints or deep within muscles. The platelet plug forms normally, explaining why bleeding time, platelet count, and PT are all normal — only the aPTT, which tests the intrinsic pathway, is prolonged.
Intrinsic Coagulation Pathway and Hemophilia A/B
X-linked Recessive Inheritance in Hemophilia
SELF-CHECK
A boy with hemophilia A has factor VIII activity of 0.8%. Which severity category does he belong to, and what type of bleeds should you anticipate?
A. Mild hemophilia (5–40%); bleeds only after major surgery
B. Moderate hemophilia (1–5%); bleeds after minor trauma
C. Severe hemophilia (<1%); spontaneous bleeds including haemarthrosis and intracranial haemorrhage
D. Moderate hemophilia (1–5%); no haemarthrosis expected
Reveal Answer
Answer: C. Severe hemophilia (<1%); spontaneous bleeds including haemarthrosis and intracranial haemorrhage
Factor VIII activity of 0.8% places him in the severe category (< 1%). Severe hemophilia is characterised by spontaneous bleeding episodes occurring without identifiable trauma, including haemarthrosis into major joints (knee, ankle, elbow), deep muscle haematomas, and the most dangerous complication — intracranial haemorrhage. Moderate hemophilia (1–5%) generally requires minor trauma to trigger bleeds; mild (5–40%) requires significant trauma or surgery.
Diagnosis and Laboratory Investigation
The laboratory diagnosis of hemophilia follows a logical two-stage approach: first establish that the bleeding phenotype is a coagulation (not platelet) disorder, then identify the specific factor deficiency. The characteristic coagulation profile — which every clinician must recognise — is a prolonged aPTT with a completely normal prothrombin time (PT), normal thrombin time (TT), normal platelet count, and normal bleeding time. This pattern uniquely identifies an isolated intrinsic pathway defect, narrowing the differential to factors XII, XI, IX, or VIII. In a male child with joint bleeds and a family history (or de novo), the most likely diagnoses are hemophilia A (factor VIII) and hemophilia B (factor IX).
The mixing test is a useful confirmatory step: mixing the patient's plasma 1:1 with normal pooled plasma will correct the prolonged aPTT in factor deficiency (as the normal plasma supplies the missing factor), but will fail to correct if the patient has developed a factor inhibitor (antibody against factor VIII or IX). This distinction is clinically critical because the management of hemophilia with inhibitors is significantly more complex.
Specific factor assays (factor VIII chromogenic or one-stage assay; factor IX one-stage assay) confirm the diagnosis and quantify severity. Inhibitor screening (Bethesda assay) should be performed in any patient who fails to respond normally to factor replacement — inhibitors develop in approximately 25–30% of patients with severe hemophilia A after repeated factor infusions and represent a major management challenge.
Imaging is used to assess the musculoskeletal consequences: X-rays and MRI of target joints evaluate the degree of haemophilic arthropathy. Ultrasound can detect muscle haematomas not clinically apparent.
| Test | Result in Hemophilia A/B | Reason |
|---|---|---|
| aPTT | Prolonged (may be 2–3× normal) | Deficient intrinsic pathway (FVIII or FIX) |
| PT | Normal | Extrinsic pathway intact |
| Platelet count | Normal | Megakaryopoiesis unaffected |
| Bleeding time | Normal | Primary haemostasis (platelet plug) intact |
| FVIII assay | Low in Hemophilia A | Confirms specific deficiency |
| FIX assay | Low in Hemophilia B | Confirms specific deficiency |
| Mixing test | Corrects aPTT | Factor deficiency (not inhibitor) |
| Inhibitor (Bethesda) | Positive if inhibitor present | Antibody neutralises replacement factor |
CLINICAL PEARL
Never give an intramuscular injection to a child with known or suspected hemophilia. An IM injection into a haemophiliac deposits drugs into a space where bleeding cannot be contained — the resulting muscle haematoma can expand silently, compress neurovascular structures, and cause acute compartment syndrome. Even routine vaccinations must be given subcutaneously (SC) with firm pressure at the injection site for at least 5 minutes. This rule extends to aspirin and NSAIDs, which impair platelet function and amplify the bleeding risk by removing the one haemostatic mechanism (primary haemostasis) that is still working.