Page 4 of 11
PE2.3 | Short Stature — SDL Guide
Learning Objectives
- Define short stature using WHO/IAP height-for-age growth chart criteria and state normal growth velocity benchmarks for prepubertal children
- Classify short stature into normal variants (familial short stature, CDGP) and pathological causes (endocrine, systemic disease, nutritional, skeletal dysplasia, chromosomal/syndromal), explaining the pathophysiological mechanism for each
- Conduct a systematic assessment of a child with short stature including mid-parental height calculation, growth velocity, bone age X-ray, and appropriate investigations
- Outline management of short stature by cause, including GH therapy indications, levothyroxine for hypothyroidism, nutritional rehabilitation, Turner syndrome management, and reassurance for normal variants
INSTRUCTIONS
Short stature is one of the most frequent reasons for referral to paediatric endocrinology, yet the majority of children referred have normal variants — familial short stature or constitutional delay — that require reassurance rather than treatment. The ability to distinguish normal variants from pathological causes, to conduct a systematic assessment, and to apply appropriate management is an essential clinical skill for every graduate. This module follows the clinical arc from presentation through to a cause-specific management approach.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 7 (Growth and Development) and Ch. 18 (Endocrine Disorders) (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 572 (Short Stature) (textbook)
- IAP 2015 Growth Charts for Indian Children (0–18 years) (guideline)
- IAP Guidelines on Growth Hormone Therapy in Children (2019) (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 10-year-old boy is brought to the outpatient clinic by his parents, who are worried that he is 'the shortest in his class.' His height is 122 cm, which plots at −2.5 SD on the IAP height-for-age chart. His weight is appropriate for his height. His father recalls being a 'late bloomer' who grew significantly after his classmates. The boy's growth velocity over the past year was 4.2 cm. His bone age X-ray reports a skeletal age of 8 years. His thyroid function and IGF-1 levels come back as normal. What is the most likely cause of his short stature, and how do you counsel the family?
WHY THIS MATTERS
Short stature is among the most anxiety-provoking findings for families and one of the top five referral reasons to paediatric outpatient clinics. In India, where stunting affects approximately 35% of children under five (NFHS-5, 2019–21), the clinician must distinguish between the very common normal variants that need only reassurance and the subset of treatable pathological causes — where early diagnosis can meaningfully change a child's final adult height. Missing growth hormone deficiency, hypothyroidism, or Turner syndrome results in preventable permanent height loss. Conversely, over-investigating every short child wastes resources and creates unnecessary parental anxiety. The skill is in systematic, cost-effective evaluation.
RECALL
Recall these foundational concepts:
• Growth hormone (GH) axis: hypothalamus secretes GHRH → stimulates anterior pituitary to release GH → GH stimulates liver (and growth plate) to produce IGF-1 (Insulin-like Growth Factor-1) → IGF-1 drives chondrocyte proliferation at the epiphyseal growth plate → linear bone growth.
• Thyroid hormone is essential for normal bone maturation; hypothyroidism causes delayed bone age and impaired linear growth.
• Bone age reflects skeletal maturation, estimated from a left-hand and wrist X-ray using the Greulich-Pyle (GP) atlas; bone age < chronological age = delayed maturation.
• Normal prepubertal growth velocity: approximately 5–7 cm/yr; values persistently below 4 cm/yr are a red flag regardless of current height.
• Mid-parental height (MPH): the genetic target height calculated as: for boys = (father's height + mother's height + 13) ÷ 2; for girls = (father's height + mother's height − 13) ÷ 2. Target range = MPH ± 8.5 cm (approximately ±2 SD).
Presenting the Problem: What is Short Stature?
Short stature is defined as a height-for-age below −2 SD from the mean for age and sex on the WHO or IAP growth chart. This corresponds approximately to the 2.3rd centile — by definition, 2.3% of a healthy population will fall below this threshold, so the mere presence of short stature does not imply pathology; context, trajectory, and cause are what matter. Severe short stature is defined as height-for-age below −3 SD, which is more likely to have an identifiable cause. The definition is population-specific: for children aged 0–5 years, the WHO 2006 multicentre growth standard is the reference; for older Indian children (5–18 years), the IAP 2015 growth chart is the preferred reference.
Growth is not a single measurement but a process; its most important metric is growth velocity — the rate of linear growth over a defined period (usually 6–12 months). A normal prepubertal growth velocity is approximately 5–7 cm/yr; a growth velocity consistently below 4 cm/yr is a red flag for pathological short stature regardless of the child's current height centile. A child at the 25th centile who is growing at only 2 cm/yr is more concerning than a child at the −2.5 SD mark who is growing at 5.5 cm/yr.
Short stature accounts for a significant proportion of paediatric endocrine consultations. In India, the background prevalence of stunting (height-for-age < −2 SD attributable to chronic undernutrition) means that the commonest cause of short stature nationally is nutritional — a fundamentally different problem from the endocrine causes seen more often in referred hospital populations. This distinction in epidemiology shapes the assessment approach: in primary care, nutrition and recurrent infections are the priority; in tertiary referral, hormonal and genetic causes rise in relative frequency.
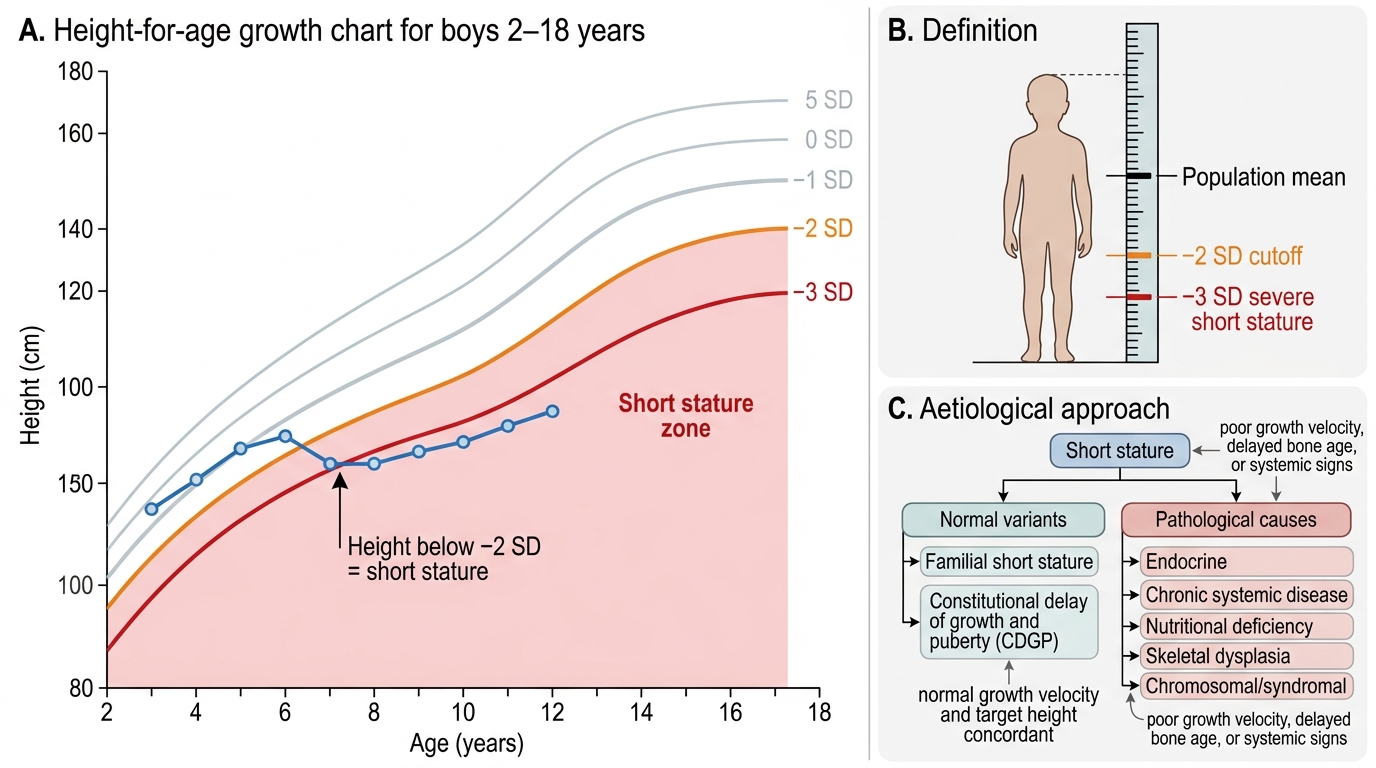
Short Stature on Height-for-Age Growth Chart
Pathophysiology and Aetiology of Short Stature
Short stature is classified into normal variants — conditions where the cause is constitutional or genetic and the child will reach a height within their genetic target range — and pathological causes, where an underlying medical problem impairs linear growth and intervention is required. Making this distinction is the core clinical task.
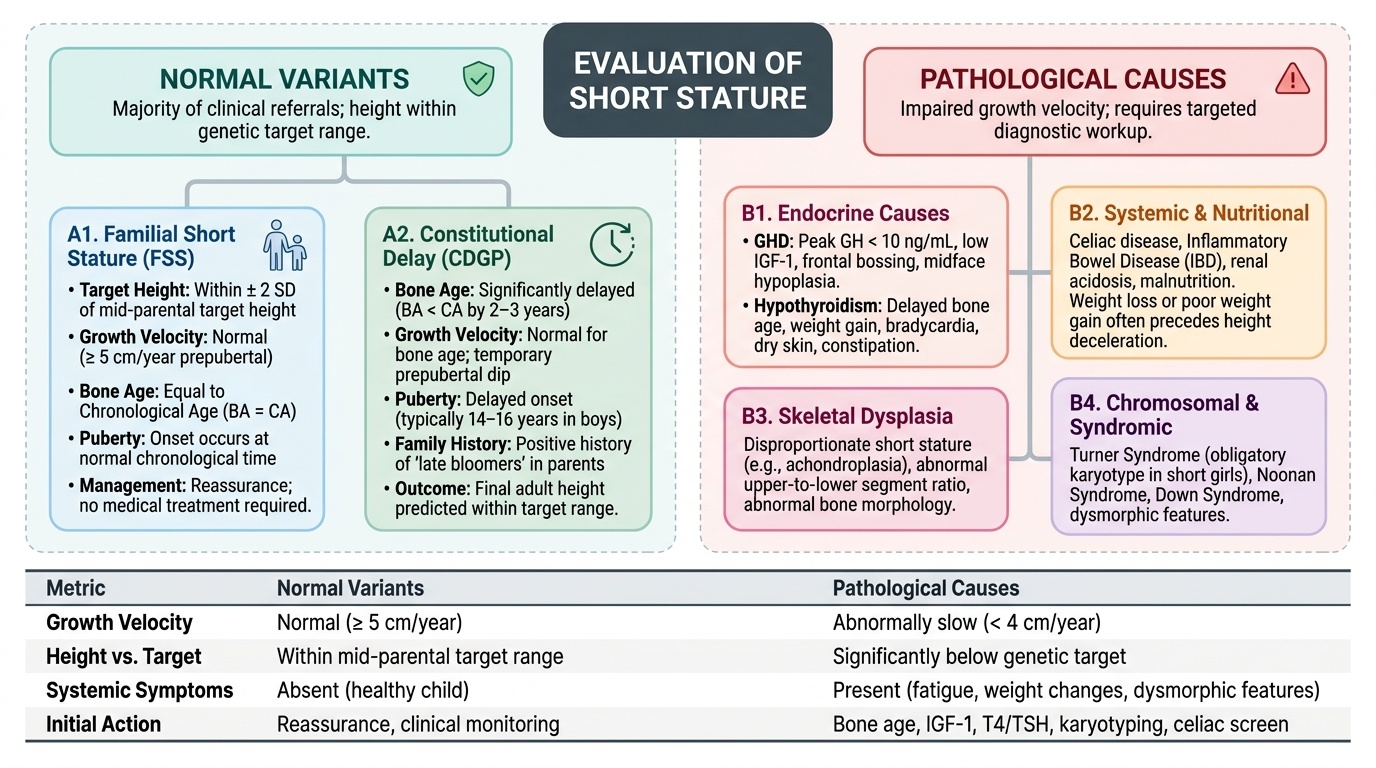
Provided image
Normal variants (together accounting for the majority of referred cases):
Familial short stature (FSS): The child is short because their parents are short; height is within ±2 SD of the mid-parental height target. Growth velocity is normal (≥5 cm/yr prepubertal), bone age equals chronological age, puberty begins at the normal time, and the child will reach a height concordant with the family pattern. No investigation or treatment is needed beyond reassurance.
Constitutional delay of growth and puberty (CDGP): The child's biological clock is delayed — bone age is significantly less than chronological age (typically 2–3 years behind), growth velocity is normal for bone age, puberty is delayed (typically beginning at 14–16 years in boys), and there is often a family history of delayed puberty in a parent ('I was a late bloomer'). Final adult height is predicted to be within the target range — the child simply reaches it later. CDGP is more common in boys and is the most frequent cause of short stature in prepubertal boys referred to endocrinology.
Pathological causes:
Endocrine causes: Growth hormone deficiency (GHD) results from inadequate GH secretion (isolated or as part of panhypopituitarism). The child has short stature, growth velocity below 4 cm/yr, delayed bone age, and characteristic physical features (frontal bossing, midface hypoplasia, high-pitched voice, increased body fat with decreased muscle mass). IGF-1 and IGFBP-3 are low. GH stimulation testing (peak GH <10 ng/mL after pharmacological stimulation) is required for diagnosis, and MRI of the pituitary is essential to exclude a craniopharyngioma or other structural lesion.
Hypothyroidism: Whether congenital (missed on neonatal screening) or acquired (autoimmune thyroiditis — Hashimoto's), hypothyroidism profoundly impairs linear growth through its effects on bone maturation and GH axis sensitivity. Delayed bone age, short stature, weight gain, constipation, bradycardia, dry skin, and poor school performance are the clinical clues. TSH is elevated; free T4 is low. Treatment with levothyroxine restores growth velocity within months.
Systemic disease: Any chronic illness consuming energy or causing malabsorption can impair growth — coeliac disease (most commonly missed), inflammatory bowel disease, chronic kidney disease (metabolic acidosis inhibits growth), cyanotic congenital heart disease, and recurrent severe infections (HIV, TB). The growth failure in these conditions is mediated by nutritional insufficiency, inflammatory cytokines (IL-6, TNF-α), and GH-IGF-1 axis suppression.
Nutritional: Protein-energy malnutrition and specific micronutrient deficiencies (zinc deficiency is particularly important for linear growth and common in India) cause stunting. This is the most prevalent cause of short stature nationally.
Skeletal dysplasias: Achondroplasia (FGFR3 gain-of-function mutation), hypochondroplasia, and other chondrodystrophies produce disproportionate short stature (sitting height:span ratio abnormal; limbs short relative to trunk). Radiological skeletal survey is diagnostic.
Chromosomal and syndromal: Turner syndrome (45,X or mosaic) presents in girls as short stature + gonadal dysgenesis (primary amenorrhoea) + characteristic features (webbed neck, wide carrying angle, shield chest, low hairline, widely spaced nipples). Down syndrome (Trisomy 21), Noonan syndrome, Prader-Willi syndrome, and Silver-Russell syndrome each have specific growth-plus-dysmorphic phenotypes. Karyotype is essential in any girl with unexplained short stature.
Intrauterine growth restriction (IUGR)/Small for gestational age (SGA): Children born SGA (<−2 SD birth weight for gestational age) who do not achieve catch-up growth by 2–4 years may have persistent short stature; approximately 10–15% of SGA children do not catch up spontaneously and may be eligible for GH therapy.
Psychosocial deprivation: Severe emotional deprivation (psychosocial dwarfism) can suppress GH secretion through hypothalamic mechanisms, producing a reversible form of GHD that normalises when the child is placed in a nurturing environment.
SELF-CHECK
A 12-year-old boy has height −2.5 SD for age, growth velocity of 5.2 cm/yr, bone age of 10 years, and his father had delayed puberty. Thyroid function and IGF-1 are normal. What is the MOST likely diagnosis?
A. Growth hormone deficiency
B. Familial short stature
C. Constitutional delay of growth and puberty (CDGP)
D. Hypothyroidism
Reveal Answer
Answer: C. Constitutional delay of growth and puberty (CDGP)
The triad of short stature + delayed bone age (10 yr in a 12-yr-old) + normal growth velocity for bone age + family history of delayed puberty is classic for constitutional delay of growth and puberty (CDGP). The normal IGF-1 and thyroid function exclude GHD and hypothyroidism. Familial short stature would have a bone age equal to chronological age and a family history of short — not delayed — adults. In CDGP the child will eventually reach their genetic target height, just later than peers.
Systematic Assessment of a Child with Short Stature
A systematic assessment of short stature requires integrating growth data, family history, physical examination, and targeted investigations to distinguish the treatable pathological causes from normal variants. The approach must be stepwise and cost-effective — not a blanket investigation panel for every short child.
Step 1 — Growth chart and growth velocity. Plot height and weight on the IAP/WHO chart. Compute height-for-age SDS. If prior growth records exist, calculate growth velocity (height gain ÷ time in years). A growth velocity below 4 cm/yr in a prepubertal child mandates investigation regardless of current height centile. Falling across centile lines (especially ≥2 centile bands) is a red flag even in a child not yet below −2 SD.
Step 2 — Mid-parental height (MPH). Calculate the genetic target height and target range (MPH ± 8.5 cm). A child whose height is well below the target range has a more likely pathological cause than one within the range. If height is within the target range and growth velocity is normal, FSS is likely.
Step 3 — Bone age X-ray (left wrist). Compare bone age (from Greulich-Pyle atlas) to chronological age. Bone age significantly less than chronological age with normal growth velocity = CDGP or GHD; bone age equal to chronological age = FSS or normal. Bone age greatly delayed beyond what CDGP would explain, or with additional features, prompts further investigation.
Step 4 — Body proportions. Measure sitting height and calculate the sitting height:height ratio (or sitting height:leg length). A ratio outside the normal range indicates disproportionate short stature → skeletal dysplasia. Measure upper:lower segment ratio. Measure arm span — a significantly reduced span relative to height suggests shortened limbs.
Step 5 — History. Birth weight and gestational age (SGA/IUGR?); neonatal history (prolonged jaundice → congenital hypothyroidism?); dietary history (nutritional short stature); symptoms of systemic disease (diarrhoea, chronic cough, exercise intolerance); developmental and school performance (hypothyroidism, chromosomal); family heights and pubertal timing history.
Step 6 — Physical examination. Dysmorphic features (Turner: webbed neck, wide carrying angle, shield chest, low hairline; Down: characteristic facies; Noonan: similar Turner-like features in boys; achondroplasia: macrocephaly, trident hand, shortened limbs). Signs of hypothyroidism (bradycardia, dry skin, delayed reflexes, goitre). Signs of Cushing's (central obesity, striae, hypertension — always ask about exogenous steroid use). Pubertal stage by Tanner staging.
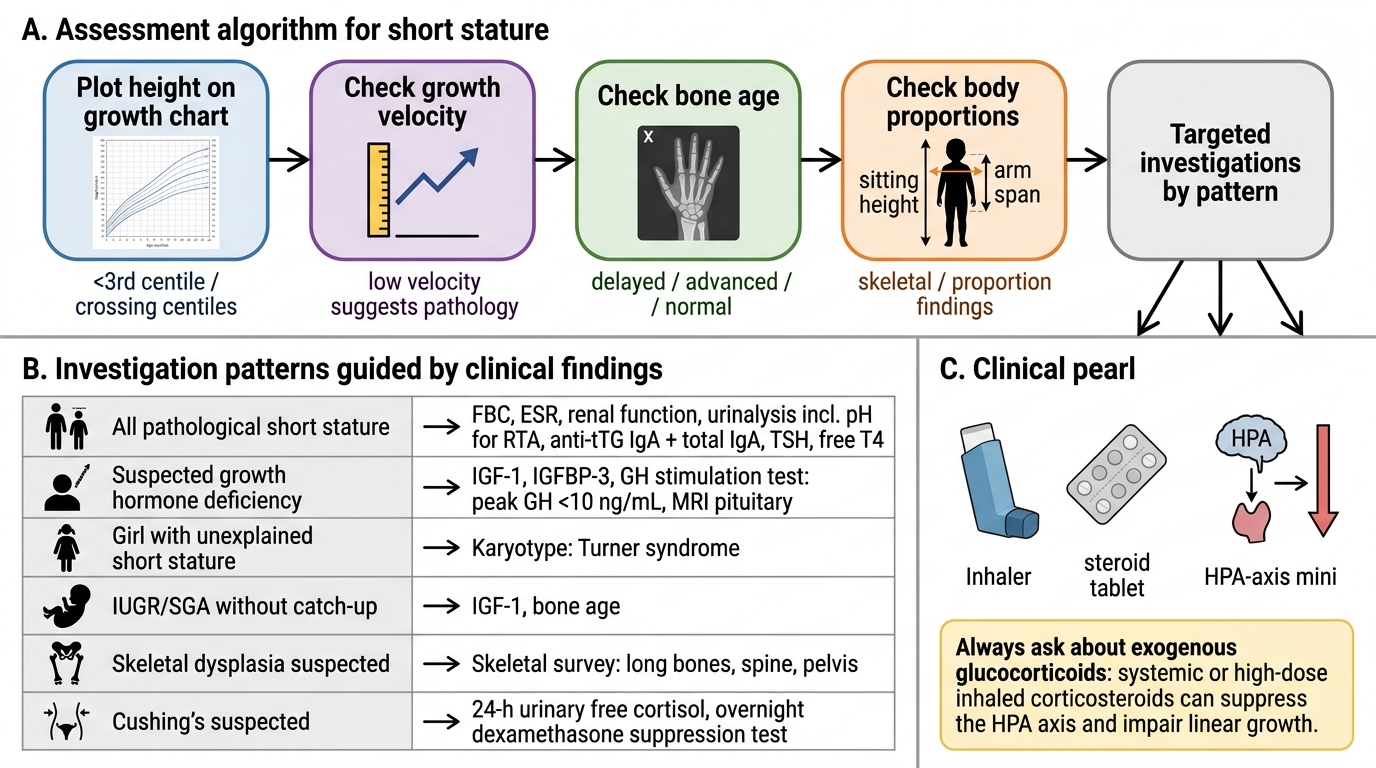
Assessment Algorithm for Short Stature
Investigations guided by clinical findings:
| Clinical finding | Investigation |
|---|---|
| All children with pathological short stature | Full blood count, ESR, renal function, urinalysis (pH for RTA), coeliac antibodies (anti-tTG IgA + total IgA), thyroid function (TSH, free T4) |
| Suspected GHD | IGF-1, IGFBP-3 (screening); GH stimulation test (peak GH <10 ng/mL = deficient); MRI pituitary |
| Girl with unexplained short stature | Karyotype (Turner syndrome) |
| IUGR/SGA without catch-up | IGF-1, bone age |
| Skeletal dysplasia suspected | Skeletal survey (X-ray long bones, spine, pelvis) |
| Cushing's suspected | 24-h urinary free cortisol, overnight dexamethasone suppression test |
CLINICAL PEARL
Always check for exogenous glucocorticoid use as a cause of short stature. Systemic or high-dose inhaled corticosteroids suppress the HPA axis and impair linear growth — yet parents frequently do not volunteer this history unless specifically asked ('Is your child on any regular medications, including inhalers, nasal sprays, or creams?'). Cushing's syndrome from steroid use presents with short stature PLUS weight gain and central redistribution of fat — a combination that distinguishes it from most other causes where height and weight often track together or weight is also low.