Page 5 of 11
PE2.3 | Short Stature — SDL Guide (Part 2)
Management of Short Stature
Management of short stature is fundamentally cause-directed. The key principle is that treating the underlying condition in pathological short stature restores growth velocity; where no treatable cause exists, the child and family require evidence-based reassurance, not placebo interventions.
Provided image
Normal variants — FSS and CDGP:
No specific treatment is required. Management is reassurance: explain that the child is growing normally for their genetic potential (FSS) or will achieve full growth potential with a delay (CDGP). For CDGP, projected final adult height should be explained in terms of the MPH target range. In adolescent boys with significant psychosocial distress from delayed puberty and short stature, a short course of low-dose testosterone (50–100 mg IM monthly for 3–6 months) can induce puberty without significantly compromising final adult height; however, this requires careful endocrinological assessment and counselling.
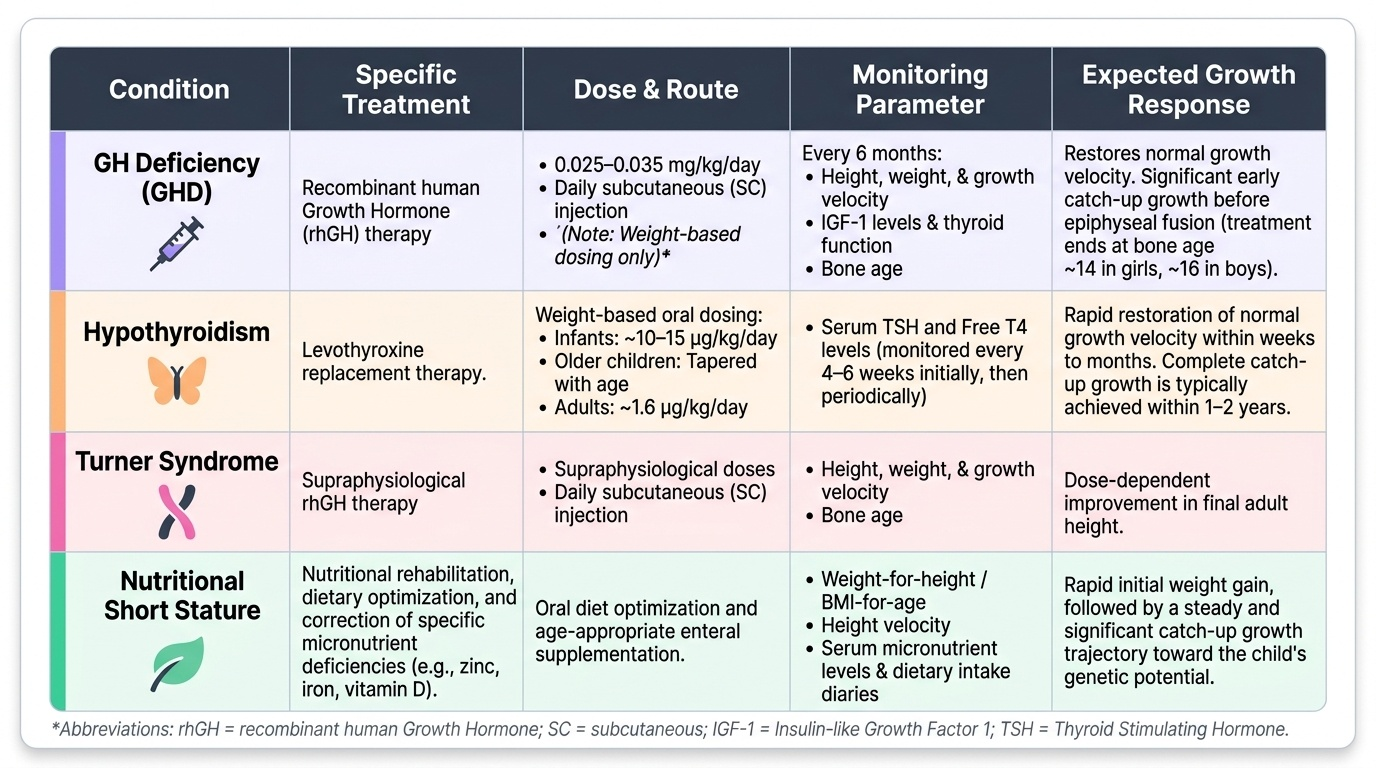
Growth hormone deficiency:
Recombinant human GH (rhGH) therapy is the standard treatment, given as a daily subcutaneous injection. The dose is 0.025–0.035 mg/kg/day (weight-based — never quote a fixed adult dose for a paediatric patient). The response is best when GHD is diagnosed and treated early, before significant bone-age advancement. Regular 6-monthly monitoring of height, weight, growth velocity, IGF-1 levels, bone age, and thyroid function is required. Adverse effects include benign intracranial hypertension (headache, papilloedema — rare), glucose intolerance, and slipped capital femoral epiphysis. Treatment continues until bone age reaches approximately 14 years in girls and 16 years in boys (epiphyses fused), or until adult height is achieved.
Additional approved indications for rhGH therapy (IAP 2019 guidelines): Turner syndrome (supraphysiological dose required), IUGR/SGA without catch-up growth by age 4 years, chronic renal insufficiency, Prader-Willi syndrome, SHOX gene deficiency, Noonan syndrome, and idiopathic short stature in select cases (final adult height gain expected ≥1.5 SD).
Hypothyroidism:
Levothyroxine replacement restores normal growth velocity within weeks to months. The dose is weight-based (~10–15 µg/kg/day in infants, tapering with age; adult dose 1.6 µg/kg/day); monitor TSH and free T4 every 4–6 weeks initially. Catch-up growth is usually complete within 1–2 years provided treatment is initiated before significant epiphyseal fusion.
Turner syndrome:
The management is multidisciplinary: rhGH therapy (at higher doses than GHD) to improve final adult height, oestrogen replacement from approximately 11–12 years to induce puberty, progestin added after 1–2 years to establish menstrual cycles. Cardiac (coarctation, bicuspid aortic valve) and renal anomalies require annual assessment. Psychological support and fertility counselling are integral.
Nutritional short stature:
Address the dietary deficiency: protein-energy rehabilitation, zinc supplementation (zinc 10–20 mg/day for 3 months is recommended by IAP for children with growth faltering), micronutrient repletion (iron, vitamin D). Growth velocity improves significantly with adequate nutrition, though full catch-up may be limited if stunting is long-standing (epiphyseal growth potential diminishes with age).
Systemic disease:
Treat the underlying condition — gluten-free diet for coeliac disease, IBD therapy, cardiac surgery for haemodynamically significant CHD. Growth velocity recovers significantly once disease is controlled.
SELF-CHECK
A 7-year-old girl has height −3 SD for age, growth velocity 2.5 cm/yr, delayed bone age, and low IGF-1. MRI pituitary shows a small adenohypophysis. She is started on recombinant human GH. What is the CORRECT weight-based dosing approach?
A. A fixed daily dose of 2 IU regardless of weight
B. 0.025–0.035 mg/kg/day as a daily subcutaneous injection
C. 0.1 mg/kg three times per week intramuscularly
D. GH dose is based on bone age, not weight
Reveal Answer
Answer: B. 0.025–0.035 mg/kg/day as a daily subcutaneous injection
Recombinant human GH for GH deficiency is dosed at 0.025–0.035 mg/kg/day as a daily subcutaneous injection. Paediatric dosing is always weight-based (mg/kg) — never a fixed adult dose. Three-times-weekly IM administration is not the current standard; daily SC injection is recommended to mimic the physiological pulsatile pattern. Dosing is based on body weight, not bone age.
Self-Assessment: Short Stature
This self-assessment section draws together the full clinical arc of short stature management, from the initial definition through to cause-specific treatment. Working through each item below will reveal whether you can apply the key concepts in a clinical reasoning context, not just recall isolated facts. The ability to reason from growth chart data to a differential diagnosis and then to a targeted investigation plan is the core skill this module aims to develop. Before attempting the quiz items, review the essential distinguishing features between the two normal variants (FSS vs CDGP) and the three most clinically important pathological causes (GHD, hypothyroidism, Turner syndrome) — these are the scenarios most likely to appear in examinations and in practice.
Must-know facts checklist:
• Short stature definition: height-for-age < −2 SD on IAP/WHO chart; severe = < −3 SD.
• Growth velocity < 4 cm/yr prepubertal = red flag regardless of current height centile.
• FSS: bone age = chronological age, growth velocity normal, height within MPH target range.
• CDGP: bone age delayed, growth velocity normal for bone age, family history of delayed puberty, normal final adult height.
• GHD: growth velocity <4 cm/yr, delayed bone age, low IGF-1, peak GH <10 ng/mL on stimulation test; treat with rhGH 0.025–0.035 mg/kg/day SC daily.
• Hypothyroidism: TSH elevated, free T4 low, delayed bone age; treat with levothyroxine (weight-based dosing).
• Turner syndrome (45,X): unexplained short stature in girls + gonadal dysgenesis; karyotype essential; treat with high-dose rhGH + oestrogen replacement.
• Mid-parental height formula: boys = (father + mother + 13)/2; girls = (father + mother − 13)/2; target range ± 8.5 cm.
• Always check for exogenous steroid use (short stature + weight gain = Cushing's).
Self-assessment prompts:
1. A 9-year-old girl presents with height −2.5 SD, weight normal-for-height, bone age of 7 years, and low-normal IGF-1. Karyotype is 45,X. What is the most important next investigation and why?
2. Why is growth velocity more useful than a single height measurement in deciding whether to investigate short stature?
3. Explain in one sentence why CDGP does NOT require GH therapy.
SELF-CHECK
A 13-year-old girl is referred for short stature (height −2.8 SD). She has had no menarche, a wide carrying angle, and a low posterior hairline. Which investigation is MOST important to perform FIRST?
A. GH stimulation test
B. Karyotype
C. MRI pituitary
D. 24-hour urinary cortisol
Reveal Answer
Answer: B. Karyotype
The combination of short stature in a girl + delayed puberty (no menarche at 13) + wide carrying angle + low hairline is the classic presentation of Turner syndrome (45,X). The single most important first investigation is a karyotype. Turner syndrome can present without all features and must be excluded in any girl with unexplained short stature. A GH stimulation test or MRI pituitary is premature before establishing the genetic diagnosis. Urinary cortisol is for Cushing's syndrome, which typically presents with weight gain rather than weight-appropriate short stature.