Page 22 of 48
PE27.7 | Epilepsy — SDL Guide
Learning Objectives
- Define epilepsy using the ILAE 2014 operational definition
- Classify seizure types and epilepsy syndromes using the ILAE 2017 framework
- Explain the pathogenesis of epilepsy — genetic, structural, metabolic, and unknown causes
- Select appropriate anti-epileptic drugs based on seizure and epilepsy type in children
INSTRUCTIONS
Epilepsy affects approximately 50–70 per 100,000 children worldwide and is the most common serious neurological condition of childhood. Unlike febrile seizures — which are age-limited and usually benign — epilepsy imposes a chronic management burden on the child and family, with implications for education, driving, employment, and mental health. This module will equip you to diagnose epilepsy precisely, classify seizure types and syndromes (the foundation of rational AED selection), and initiate appropriate treatment in a paediatric setting.
References
- Ghai Essential Pediatrics, 9th ed., Ch 18 (Neurology — Epilepsy) (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch 609 (Seizures in Childhood) (textbook)
- ILAE 2017 Operational Classification of Seizure Types (Fisher et al., Epilepsia) (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 7-year-old girl is referred by her school teacher who has noticed that she 'switches off' multiple times a day for 5–10 seconds — staring blankly, not responding when called, then resuming activity as if nothing happened. Her teachers first thought she was daydreaming, but they now note it happens 20–30 times a day, sometimes mid-sentence. She has no recollection of the episodes. Her parents report no convulsions. EEG shows bursts of 3 Hz generalised spike-and-wave discharges coinciding with hyperventilation. This is childhood absence epilepsy (CAE) — one of the classic paediatric epilepsy syndromes, and a perfect teaching case for the ILAE classification system. Her seizure type (typical absence) and syndrome (CAE) together determine the ideal anti-epileptic drug — and critically, which drug to avoid.
WHY THIS MATTERS
Epilepsy affects 50–70 per 100,000 children and is the most common serious chronic neurological condition in paediatric practice. It is not a single disease — it is a spectrum of disorders with very different aetiologies, seizure types, prognoses, and treatment approaches. The single most important clinical skill in epilepsy management is accurate seizure classification: choosing the wrong anti-epileptic drug because of misclassification causes seizure aggravation (carbamazepine worsening absence and myoclonic seizures is a classic iatrogenic error), while correct classification aligns treatment with pathophysiology. As a final-year student, you will be the first to hear a seizure history from a parent — your description of the event to a neurologist must be systematic and use the ILAE framework.
RECALL
Recall from Physiology (PY): a seizure is caused by excessive, abnormally synchronous discharge of a population of neurons. Neuronal firing depends on the balance of excitatory (glutamate, NMDA and AMPA receptor-mediated) and inhibitory (GABA, predominantly GABA-A receptor-mediated) neurotransmission. Voltage-gated sodium channels (Na⁺) control the action potential upstroke; potassium channels (K⁺) govern repolarisation. Recall from Febrile Seizures (PE27.6): febrile seizures are age-limited provoked seizures. Epilepsy differs in that the seizures are unprovoked (occur without an acute trigger) and recurrent, reflecting a persistent alteration in neuronal network excitability.
Clinical Presentation and Seizure Types
Epilepsy is defined by the International League Against Epilepsy (ILAE, 2014) as any of the following: (1) at least two unprovoked (or reflex) seizures occurring more than 24 hours apart; (2) one unprovoked seizure and a probability of further seizures of at least 60%, similar to the general recurrence risk after two unprovoked seizures; or (3) diagnosis of an epilepsy syndrome. This operational definition captures the essential feature: recurrent, self-sustaining tendency to generate seizures without an acute brain insult.
Recognising seizure type at the bedside requires a systematic witness history. The ILAE 2017 classification begins with the question: where did the seizure START in the brain — one hemisphere (focal) or both simultaneously (generalised)? This onset type then narrows down to the specific semiology and eventually to syndrome identification, which drives treatment. Key seizure types with their clinical presentations are:
Generalised seizure types:
• Tonic-clonic (GTCS) — tonic stiffening followed by rhythmic bilateral clonic jerking; postictal drowsiness lasting minutes to hours; most dramatic and frightening for parents
• Typical absence — sudden brief (5–20 second) stare with blank expression and unresponsiveness; abrupt onset and termination; no postictal phase; child may blink or show minor oral automatisms; provoked by hyperventilation in the clinic
• Myoclonic — sudden, brief, involuntary jerks; often bilateral, especially of the upper limbs; occur in clusters; morning jerks spilling milk or cereal is a classic history in Juvenile Myoclonic Epilepsy
• Tonic — sustained stiffening of limbs and trunk; common in sleep; associated with Lennox-Gastaut syndrome
• Atonic (drop attacks) — sudden loss of postural tone; child drops to floor; leads to facial/head injuries; padded helmet sometimes required; seen in Lennox-Gastaut
• Epileptic spasms (infantile spasms) — sudden brief flexion (jackknife) or extension of trunk and limbs; occur in clusters; onset 3–12 months; associated with West syndrome; a neurodevelopmental emergency
Focal seizure types:
• Focal aware (formerly 'simple partial') — preserved consciousness; focal motor (arm jerking, head deviation), sensory (tingling, visual phenomena), autonomic (nausea, pallor), or psychic (déjà vu, fear) features
• Focal impaired awareness (formerly 'complex partial') — impaired consciousness; automatisms (lip-smacking, hand fumbling, purposeless movements); postictal confusion
• Focal to bilateral tonic-clonic — begins focally then generalises; the initial focal features (aura, head deviation) provide localising information
The duration and semiology of each seizure type is clinically distinctive. Absence seizures, for instance, are easily mistaken for 'daydreaming' or attention problems — the key distinguishing feature is abrupt onset and termination, stereotyped repetition, and precipitation by hyperventilation in the consulting room. Asking a 7-year-old to blow on a pinwheel for 2–3 minutes will reliably provoke a typical absence seizure if CAE is present.
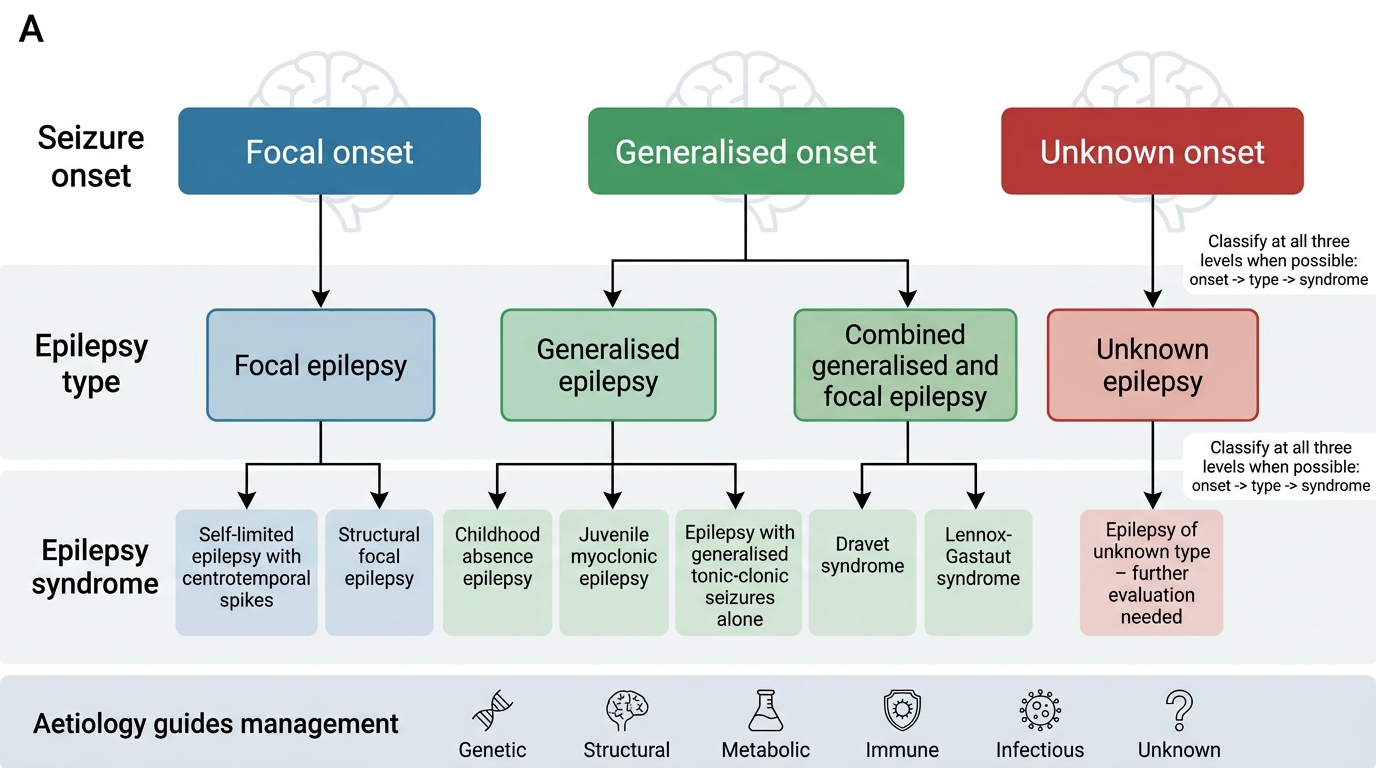
ILAE 2017 Operational Classification of Epilepsy
Pathogenesis of Epilepsy
The pathogenesis of epilepsy encompasses the mechanisms by which a normal brain is transformed into one that generates spontaneous, recurrent seizures — a process called epileptogenesis — and the mechanisms of individual seizure generation (ictogenesis). Understanding pathogenesis is essential because modern AED selection targets specific pathogenic mechanisms, and genetic epilepsies in particular require specific treatments that differ from those used in structural epilepsies. The ILAE classifies epilepsy aetiology into six categories — genetic, structural, metabolic, immune, infectious, and unknown — and identifying the aetiology directs not just AED choice but also eligibility for disease-modifying therapies (surgery, immunotherapy, dietary therapy, enzyme replacement). Importantly, aetiology and pathogenesis are not the same: the same genetic mutation (e.g., SCN1A) can cause different clinical syndromes depending on its effect on specific cell types, illustrating that the mechanism at the cellular level must be understood to select the correct treatment.
Ictogenesis — the excitation-inhibition imbalance:
Every seizure reflects an acute breakdown of the normal balance between neuronal excitation and inhibition. The following mechanisms have been identified:
1. Enhanced excitatory neurotransmission — increased glutamate (NMDA/AMPA receptor activation), increased firing of pyramidal neurons, failure of synaptic glutamate reuptake
2. Reduced inhibitory neurotransmission — reduced GABA release, GABA-A receptor dysfunction, loss of GABAergic interneurons (as in hippocampal sclerosis)
3. Abnormal voltage-gated ion channels — SCN1A (Nav1.1) loss-of-function mutants reduce Na⁺ channel activity in inhibitory interneurons, paradoxically reducing inhibition and increasing network excitability (the mechanism in Dravet syndrome)
4. Synaptic reorganisation — in chronic epilepsy, axonal sprouting (mossy fibre sprouting in the dentate gyrus) creates recurrent excitatory circuits that lower seizure threshold
Aetiology classification (ILAE 2017):
• Genetic — single-gene mutations or copy-number variants causing epilepsy syndromes; examples: SCN1A (Dravet syndrome), SCN2A, GABRG2 (GABA receptor — febrile seizures plus), KCNQ2 (neonatal epilepsy), CDKL5, ARX (West syndrome variants)
• Structural — visible brain lesion on MRI: focal cortical dysplasia (most common surgically remediable cause), hippocampal sclerosis, tuberous sclerosis complex (TSC — cortical tubers), perinatal stroke, polymicrogyria, hemimegalencephaly
• Metabolic — treatable metabolic disorders causing epilepsy: pyridoxine-dependent epilepsy (ALDH7A1 mutation — responsive to pyridoxine), glucose transporter deficiency (GLUT1 — ketogenic diet), biotinidase deficiency, phenylketonuria (PKU)
• Immune — autoimmune encephalitis with antibodies to neuronal surface antigens: anti-NMDAR encephalitis (most common — presents with behavioural change + seizures in children and young adults), LGI1, CASPR2 antibodies. Early immunotherapy (steroids, IVIG, plasma exchange) is effective.
• Infectious — ongoing CNS infection: neurocysticercosis (the most common acquired structural epilepsy cause globally, including India), herpes encephalitis, bacterial meningitis, cerebral malaria
• Unknown — no structural, metabolic, genetic, immune, or infectious cause identified despite thorough workup; the largest category in clinical practice
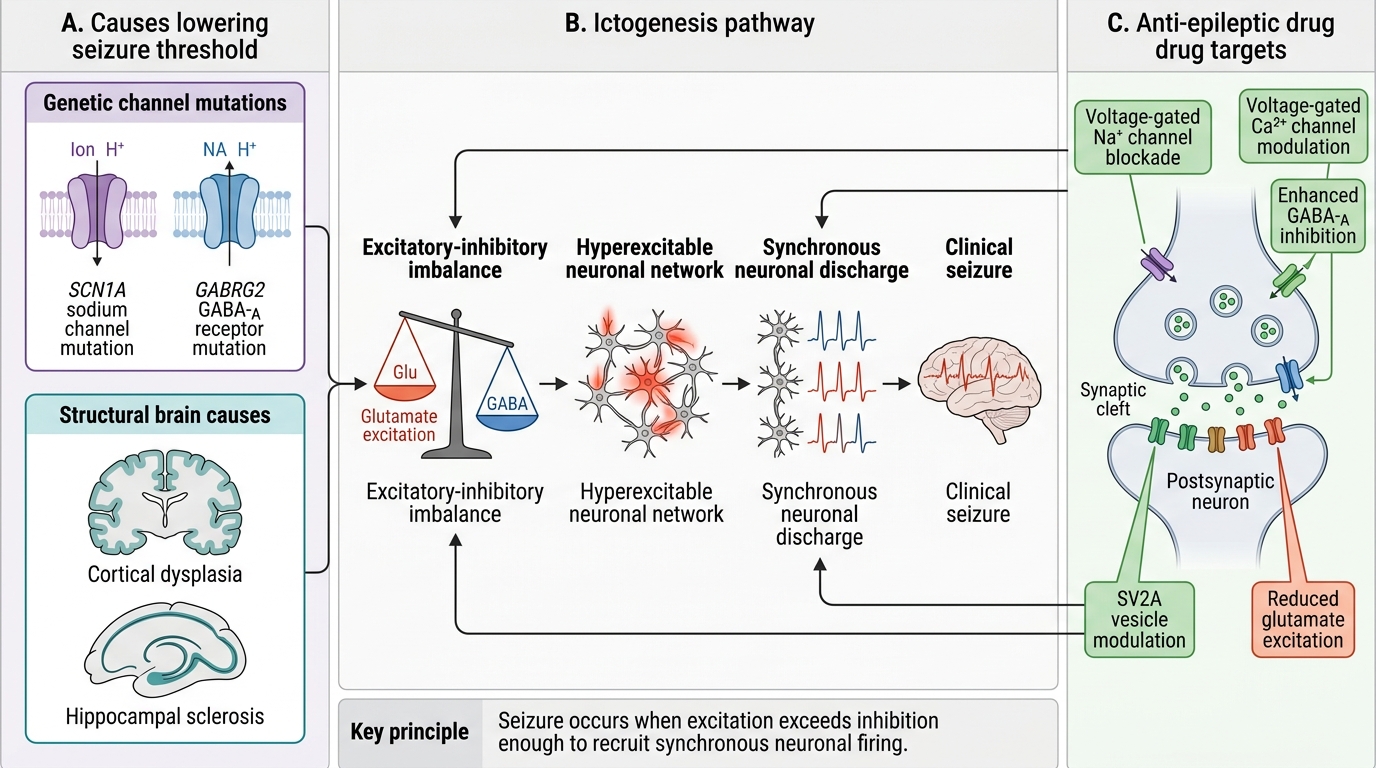
Mechanism of Ictogenesis and Anti-Epileptic Drug Targets
ILAE Classification and Epilepsy Syndromes
The ILAE 2017 operational classification has three levels: seizure type (focal/generalised/unknown onset), epilepsy type (focal, generalised, combined, unknown), and epilepsy syndrome. The syndrome level integrates age of onset, seizure type(s), EEG signature, associated features, aetiology, and prognosis — and it is at the syndrome level that treatment decisions are most precise. The following are the key paediatric epilepsy syndromes every student must know, because each has a characteristic presentation and a preferred first-line AED:
Knowing these syndromes prevents the most common prescribing errors. The most dangerous error in paediatric epilepsy prescribing is giving carbamazepine or oxcarbazepine to a child with generalised epilepsy (absence, myoclonic, or JME) — these sodium-channel blockers worsen absence and myoclonic seizures. This is not a rare event; it happens when a doctor prescribes by seizure frequency without classifying seizure type.
| Syndrome | Age of onset | Seizure type(s) | EEG | Prognosis | First-line AED |
|---|---|---|---|---|---|
| West syndrome (Infantile spasms) | 3–12 months | Epileptic spasms (clusters) | Hypsarrhythmia | Poor without early treatment; 50–70% neurodevelopmental sequelae | ACTH, vigabatrin; prednisolone |
| Childhood Absence Epilepsy (CAE) | 4–10 years | Typical absence | 3 Hz generalised spike-wave | 60–70% remission by adolescence | Ethosuximide (absence alone); valproate (absence + GTCS risk) |
| Benign Childhood Epilepsy with Centrotemporal Spikes (BECTS/Rolandic) | 5–12 years | Focal (face, tongue, arm); nocturnal GTCS | Centrotemporal spikes, normal background | Self-limiting by puberty; may not need AED | No AED often; valproate, carbamazepine if frequent |
| Juvenile Myoclonic Epilepsy (JME) | 12–18 years | Myoclonic jerks (morning) + GTCS; occasional absence | Generalised polyspike-wave | Lifelong — >90% relapse on AED withdrawal | Valproate (most effective); levetiracetam (women of reproductive age) |
| Lennox-Gastaut Syndrome (LGS) | 1–7 years | Tonic (nocturnal), atonic (drop attacks), atypical absence | Slow spike-wave <2.5 Hz; paroxysmal fast | Severe, drug-resistant; intellectual disability universal | Valproate, lamotrigine, clobazam; rufinamide, felbamate |
| Dravet syndrome (SMEI) | 1st year; febrile seizures onset | Prolonged febrile hemiclonic GTCS; later myoclonic + absence | Normal initially; later generalised/multifocal | Severe, drug-resistant; high mortality (SUDEP) | Valproate + clobazam; stiripentol; AVOID Na-channel blockers (worsen) |
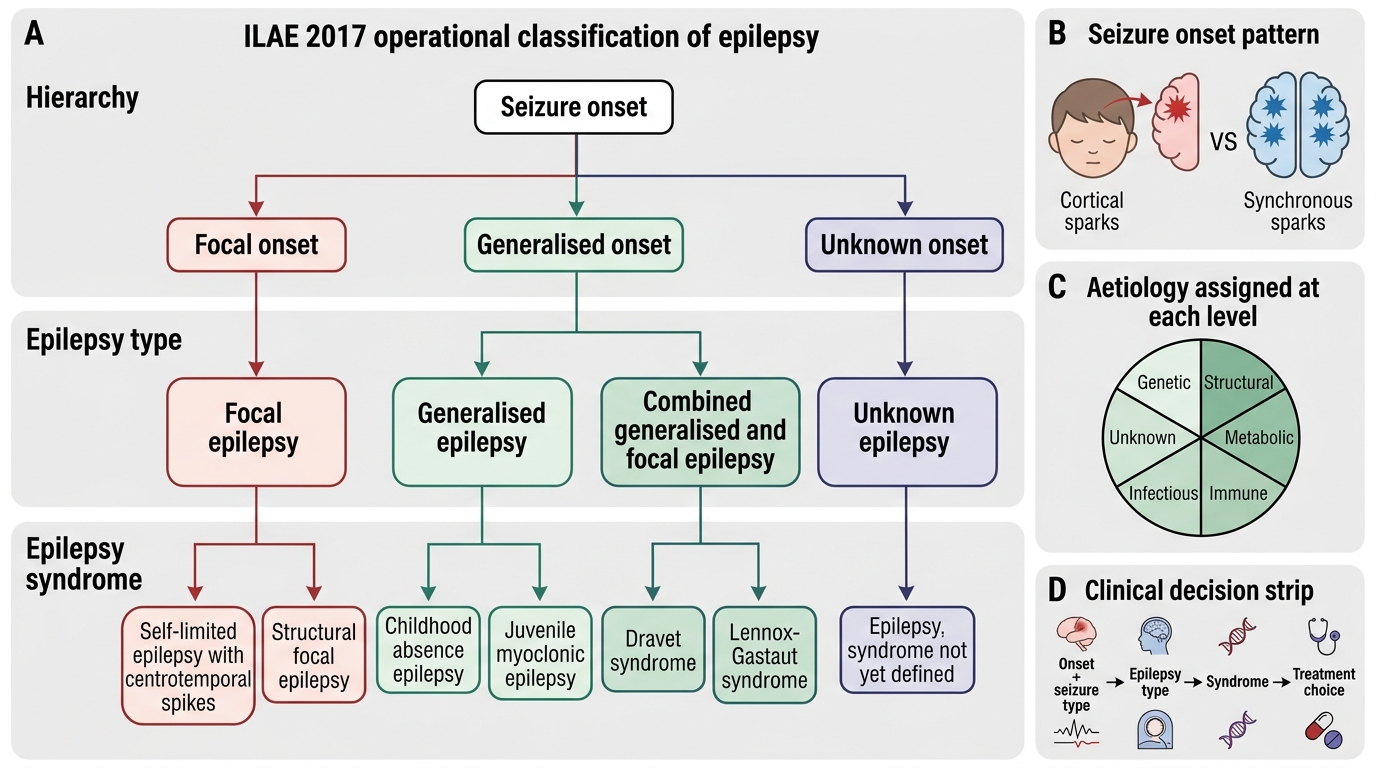
ILAE 2017 Operational Classification of Epilepsy
SELF-CHECK
A 10-year-old boy has been having brief episodes of staring, unresponsiveness, and occasional lip-smacking since age 8. Each episode lasts 10–15 seconds with no postictal phase. He is normal between episodes. EEG shows 3 Hz generalised spike-wave discharges activated by hyperventilation. Which AED is CONTRAINDICATED in this child?
A. Ethosuximide
B. Valproate
C. Carbamazepine
D. Levetiracetam
Reveal Answer
Answer: C. Carbamazepine
This child has Childhood Absence Epilepsy (CAE) — typical absence seizures with 3 Hz generalised spike-wave on EEG. Carbamazepine is a sodium-channel blocker that is effective for focal seizures but CONTRAINDICATED in generalised epilepsies including CAE. It paradoxically worsens absence and myoclonic seizures by altering thalamo-cortical synchrony. Ethosuximide (absence-specific, first-line if no GTCS risk) and valproate (broad-spectrum, first-line if GTCS risk present) are the correct first-line choices. Levetiracetam is a reasonable alternative. Prescribing carbamazepine here would cause seizure worsening.