Page 23 of 48
PE27.7 | Epilepsy — SDL Guide (Part 2)
Diagnosis and Investigation
The diagnosis of epilepsy is clinical — based on a detailed seizure history (from parent/witness and child), neurological examination, and supportive investigations. No investigation 'diagnoses' epilepsy; rather, the EEG and MRI characterise the seizure type, identify the underlying aetiology, and guide treatment decisions. The history must establish whether the paroxysmal event was truly a seizure (not a syncope, night terror, breath-holding spell, or tic) and, if a seizure, whether it was focal or generalised — information that determines both the appropriate investigation and the first-line treatment. A systematic approach to the seizure history asks: (i) What exactly happened — tonic? clonic? staring? jerking? Did it begin on one side? (ii) Were there warning symptoms (aura) before? (iii) Was consciousness lost? (iv) How long did it last? (v) Was there postictal drowsiness and for how long? This five-point history framework, elicited before any investigation is ordered, is more discriminating than any test.
Electroencephalogram (EEG) — the cornerstone investigation:
The EEG records spontaneous electrical activity of cerebral cortex via scalp electrodes. Key principles:
• A normal inter-ictal EEG does not exclude epilepsy — a single routine EEG is normal in up to 50% of children with epilepsy; repeat EEG (sleep-deprived, prolonged, or with activation procedures) increases yield
• Provocation techniques: hyperventilation (3 minutes) reliably induces typical absence; photic stimulation provokes photosensitive epilepsies (JME)
• Ictal EEG is definitive but requires the seizure to occur during recording; video-EEG (simultaneous clinical and EEG recording) is the gold standard for seizure classification and pre-surgical evaluation
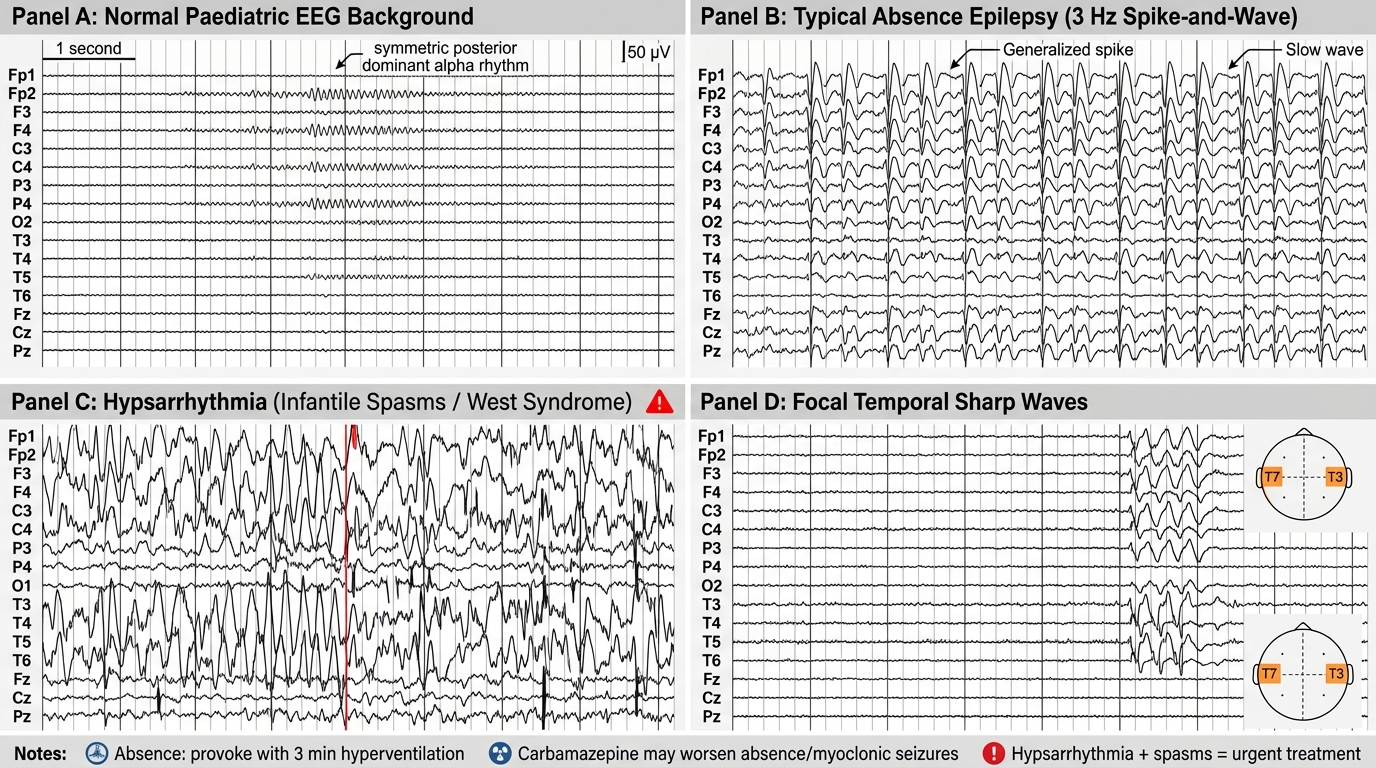
• Key EEG signatures by syndrome:
- 3 Hz generalised spike-and-wave (synchronous, bilateral) → typical absence (CAE)
- Hypsarrhythmia (chaotic, high-amplitude, multifocal spikes + slow waves with no normal background) → infantile spasms (West syndrome) — this is a neurodevelopmental emergency; diagnose and treat within days
- Centrotemporal sharp waves with normal background → BECTS/Rolandic epilepsy
- Slow (<2.5 Hz) spike-and-wave + paroxysmal fast activity (PFA) during sleep → Lennox-Gastaut
- Generalised polyspike-wave → JME (most apparent on awake or sleep-deprived EEG)
MRI brain:
• Indicated in all children with focal epilepsy, focal neurological signs, intellectual disability + seizures, drug-resistant epilepsy, or infantile spasms
• Optimal protocol: dedicated epilepsy MRI (1.5T or 3T) with FLAIR, T1 volumetric, T2, DWI, and coronal hippocampal sequences
• Yield: identifies structural aetiology in ~30% of all epilepsy, ~70% of drug-resistant focal epilepsy
• Not routinely required for straightforward CAE or BECTS (clinical-EEG diagnosis is sufficient)
Metabolic investigations:
• Glucose, electrolytes, calcium, magnesium — exclude metabolic causes, especially in neonates and infants
• Pyridoxine trial (100 mg IV/IM) in neonates with intractable seizures — pyridoxine-dependent epilepsy
• Urine organic acids, plasma amino acids, lactate, ammonia — if metabolic encephalopathy suspected
• Biotinidase enzyme assay — biotinidase deficiency (treatable)
Genetic/autoimmune testing:
• Gene panel (SCN1A, CDKL5, KCNQ2, etc.) — when a genetic syndrome is suspected (infantile spasms, Dravet, intractable neonatal seizures)
• Autoimmune antibody panel (anti-NMDAR, LGI1, CASPR2) — subacute onset seizures + encephalopathy, especially with behavioural change
EEG Patterns in Paediatric Epilepsy
CLINICAL PEARL
Carbamazepine worsens absence seizures and myoclonic jerks — prescribing it without seizure classification is one of the most common iatrogenic errors in paediatric epilepsy. A child with childhood absence epilepsy or juvenile myoclonic epilepsy given carbamazepine will experience more frequent seizures, not fewer. Always classify seizure type (focal vs generalised) before selecting an AED. The bedside test for absence seizures is asking the child to hyperventilate for 3 minutes — a typical absence is reliably provoked. This costs nothing and gives you the diagnosis at the clinic visit. A second equally important pearl: hypsarrhythmia on EEG in an infant with clustering episodes is a medical emergency — West syndrome (infantile spasms) causes permanent neurodevelopmental regression if treatment is delayed beyond a few weeks.
Management: Anti-Epileptic Drug Therapy
The goal of anti-epileptic drug (AED) therapy is seizure freedom without intolerable side effects, using the lowest effective dose of the most appropriate drug for the child's specific seizure and epilepsy type. AED selection in children is governed by three principles: (1) match the AED to the seizure/epilepsy type (mechanism matters — a drug that blocks sodium channels helps focal seizures but worsens absence/myoclonic); (2) use weight-based dosing — never a fixed adult dose; and (3) start low and titrate slowly to minimise adverse effects. Most children with newly diagnosed epilepsy should be started on monotherapy, not a combination — polypharmacy increases adverse effects without necessarily improving efficacy in newly diagnosed cases. AED therapy is initiated after a second unprovoked seizure in most cases; some guidelines allow initiation after a first seizure if the risk of recurrence is very high (e.g., structural lesion, abnormal EEG, focal neurological deficit). The SANAD II trial demonstrated that levetiracetam is non-inferior to valproate for focal epilepsy with a better tolerability profile, particularly important in women of reproductive age.
Provided image
For generalised epilepsies (absence, myoclonic, GTCS, combined):
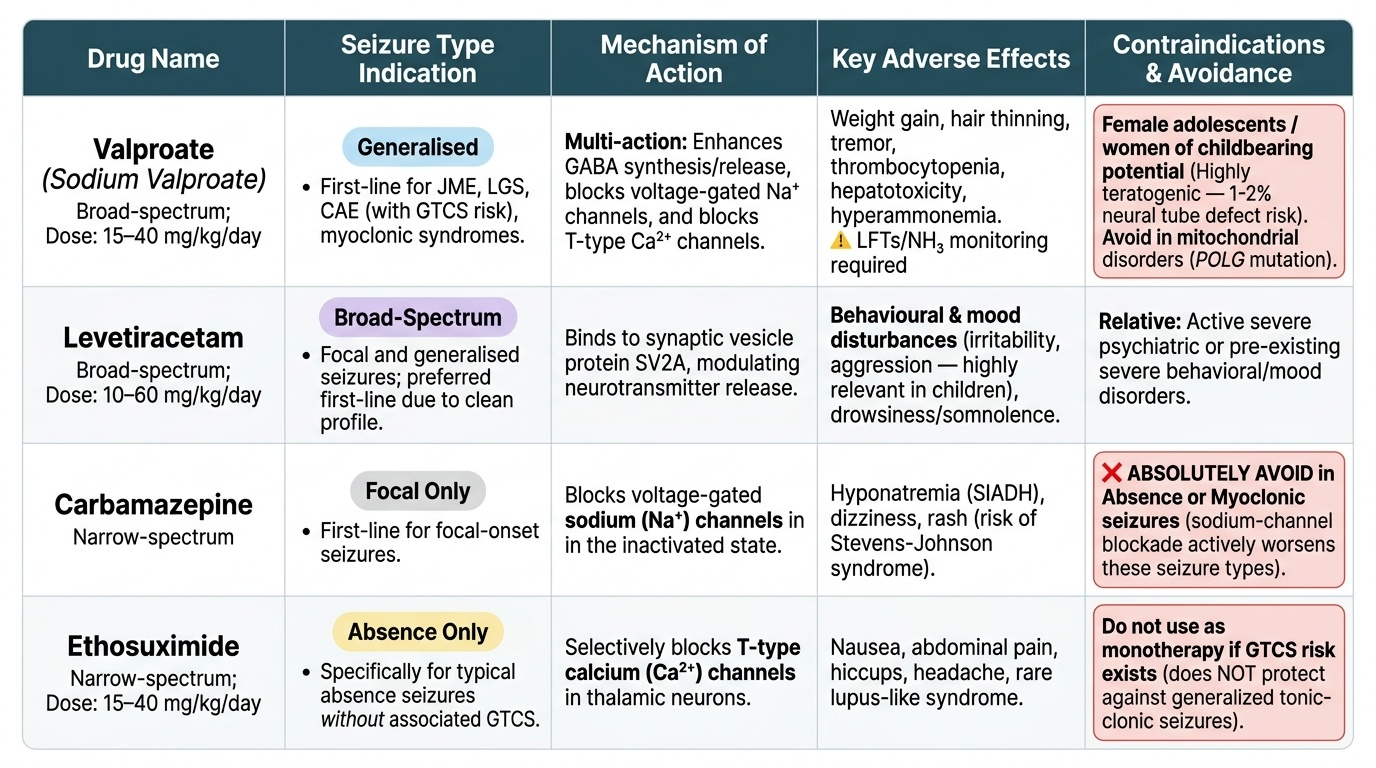
• Valproate (sodium valproate): broad-spectrum; first-line for most generalised epilepsies including JME, LGS, myoclonic syndromes, and CAE when GTCS risk present. Dose: 15–40 mg/kg/day in 2–3 divided doses. Teratogenic (neural tube defects — 1–2% risk with folic acid supplementation; higher without); avoid in adolescent females of reproductive potential if alternatives exist. Monitoring: LFTs, NH₃, full blood count (thrombocytopenia, hepatotoxicity risk).
• Ethosuximide: narrow-spectrum; specifically for typical absence seizures WITHOUT associated GTCS; does NOT protect against GTCS. Dose: 15–40 mg/kg/day. Side effects: nausea, hiccups; rare lupus-like syndrome.
• Levetiracetam: broad-spectrum; increasingly the preferred first-line for generalised and focal seizures in children (favourable pharmacokinetics, no drug interactions, no monitoring requirements). Dose: 10–60 mg/kg/day in 2 doses. Side effects: behavioural/mood disturbance (irritability, aggression — important in children), drowsiness.
• Lamotrigine: broad-spectrum; effective for focal, GTCS, absence; requires very slow titration to avoid Stevens-Johnson syndrome (SJS). Valproate doubles its serum levels — must halve the dose when co-prescribed.
For focal epilepsies:
• Carbamazepine: narrow-spectrum sodium-channel blocker; excellent for focal onset seizures. Contraindicated in absence and myoclonic epilepsies (worsens both). Dose: 5–20 mg/kg/day; monitor sodium (SIADH risk), blood count.
• Oxcarbazepine: prodrug of carbamazepine active metabolite; similar efficacy, fewer drug interactions, same contraindication (avoid in generalised epilepsies). Hyponatraemia more common.
• Levetiracetam: also effective for focal seizures.
Specific situations:
• Infantile spasms (West syndrome): ACTH + vigabatrin (first-line combination in India and UK, per UKISS); oral prednisolone as alternative. Goal: eliminate hypsarrhythmia within 2 weeks. Delayed treatment worsens neurodevelopmental outcome.
• Dravet syndrome: valproate + clobazam; stiripentol (add-on). Avoid sodium-channel blockers (carbamazepine, lamotrigine, phenytoin) — all worsen Dravet (SCN1A is a sodium channel gene; these drugs reduce its residual activity).
• Febrile seizure-plus (FS+) / early febrile seizures evolving to Dravet: SCN1A test first; if positive, avoid Na-channel blockers regardless of current seizure type.
General principles:
• Start with one drug (monotherapy); add second drug only if monotherapy at adequate dose fails
• Drug-resistant epilepsy (failure of ≥2 appropriate AEDs) — refer to specialist; consider ketogenic diet, vagus nerve stimulation, or epilepsy surgery
• All dosing is weight-based (mg/kg) in children — never use adult fixed doses
• Monitor for side effects at each visit (behaviour, growth, liver function, blood count depending on drug)
• Do NOT withdraw AEDs abruptly — taper over weeks to months to avoid withdrawal seizures
SELF-CHECK
A 15-year-old girl has had early morning jerks of her arms (especially while having breakfast) for 6 months, followed on three occasions by a generalised tonic-clonic seizure. EEG shows generalised polyspike-wave, activated by photic stimulation. She has a normal MRI. She is about to start valproate. What is the MOST IMPORTANT specific counselling point for her?
A. Valproate is not effective for her seizure type — she needs carbamazepine
B. Valproate is teratogenic and she needs reliable contraception if sexually active; consult about alternative AEDs
C. She can stop valproate after 2 years seizure-free — JME always remits
D. She does not need lifelong treatment as her seizures are mild
Reveal Answer
Answer: B. Valproate is teratogenic and she needs reliable contraception if sexually active; consult about alternative AEDs
This is Juvenile Myoclonic Epilepsy (JME) — morning myoclonic jerks + GTCS + generalised polyspike-wave on EEG (activated by photic stimulation). Valproate is the most effective first-line drug for JME. However, valproate carries a significant teratogenic risk (neural tube defects) and is a category D teratogen. For any adolescent female starting valproate, contraception counselling and a discussion about alternative AEDs (levetiracetam, lamotrigine — less effective but safer in pregnancy) is mandatory. Option C is wrong: JME is a lifelong epilepsy; >90% of patients relapse if AEDs are withdrawn, even after years of seizure freedom. Carbamazepine would worsen her JME.
Self-Assessment
Test your understanding with the following case-based questions:
Case A: An 8-month-old girl is brought with sudden clusters of jerking movements — she bends forward, throws her arms out, and cries, followed by a cluster of 10–15 similar movements over 1–2 minutes. These clusters occur 3–4 times a day. She was previously developing normally. EEG shows hypsarrhythmia.
Q1. What is the diagnosis, and what is the treatment urgency?
Answer: West syndrome (infantile spasms). This is a neurodevelopmental emergency. Treatment must be initiated within days to prevent progressive neurodevelopmental regression. First-line treatment in India: ACTH injection or oral prednisolone, combined with vigabatrin (if tuberous sclerosis complex is the aetiology, vigabatrin is particularly effective). Goal: abolish hypsarrhythmia on EEG within 2 weeks.
Case B: A 12-year-old is started on carbamazepine by a general practitioner for 'fits.' Two weeks later, his teacher reports he is 'blanking out' even more frequently. His parents notice he is also having brief upper-limb jerks in the morning.
Q2. What has happened, and what is the likely correct diagnosis and AED?
Answer: Carbamazepine has aggravated his seizures. His clinical picture (absence-type staring + morning myoclonic jerks) is consistent with Juvenile Myoclonic Epilepsy (JME) or Childhood Absence Epilepsy with evolving JME features. Carbamazepine worsens both absence and myoclonic seizures. It should be discontinued and replaced with valproate (or levetiracetam if valproate is unsuitable). Correct seizure classification BEFORE prescribing is the key lesson.
Key summary points:
• ILAE 2014 definition: ≥2 unprovoked seizures >24 h apart (or 1 seizure + ≥60% recurrence risk, or epilepsy syndrome).
• ILAE 2017 classification: focal onset → focal epilepsy; generalised onset → generalised epilepsy.
• Carbamazepine is contraindicated in absence and myoclonic epilepsies — this is the most dangerous prescribing error in paediatric epilepsy.
• Hypsarrhythmia = West syndrome = neurodevelopmental emergency — treat within days.
• AED dosing in children is ALWAYS weight-based (mg/kg).
• Valproate is teratogenic — counsel adolescent females about contraception.
• Drug-resistant epilepsy (failure of ≥2 AEDs) → refer for ketogenic diet, VNS, or surgery evaluation.