Page 1 of 14
PE29.1-3 | Down Syndrome — SDL Guide
Learning Objectives
- Describe the genetic basis of Down syndrome including the three cytogenetic mechanisms (free trisomy 21, robertsonian translocation, mosaicism) and their approximate frequencies
- Identify the characteristic clinical features and major system-wise complications of Down syndrome
- Interpret a normal karyotype and recognise trisomy 21 from a karyogram
- Outline the prenatal screening and diagnostic investigations for Down syndrome
- Formulate a comprehensive management and surveillance plan for a child with Down syndrome
- Counsel parents regarding the present child and the risk of recurrence in subsequent pregnancies, distinguishing by cytogenetic mechanism
INSTRUCTIONS
Down syndrome is the most common chromosomal disorder causing intellectual disability in children, with a birth prevalence of approximately 1 in 700–1000 live births. As a final-year student, you will encounter children with Down syndrome in paediatric wards, genetics clinics, and special schools. Beyond recognition of the phenotype, you must be able to explain the cytogenetic basis to parents, interpret a karyotype, counsel about recurrence risk (which differs critically by whether the cause is free trisomy, translocation, or mosaicism), and coordinate multidisciplinary care. This module integrates all three of these clinical skills into a single, coherent SDL.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 22 — Chromosomal Disorders (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 100 — Down Syndrome (textbook)
- American Academy of Pediatrics: Health Supervision for Children and Adolescents with Down Syndrome (2022) (guideline)
- Indian Academy of Pediatrics Guidelines on Developmental Disabilities (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 2-day-old neonate is referred to the newborn nursery for assessment. The mother, a 38-year-old primigravida, had declined antenatal screening. On examination, the infant has upslanting palpebral fissures, a flat nasal bridge, a single palmar crease bilaterally, and a generalised hypotonia. The paediatrician suspects a chromosomal abnormality and orders a karyotype. The parents ask: 'Will our baby ever go to school? What happened to cause this? Could it happen again?' You must be able to answer these questions confidently.
WHY THIS MATTERS
Down syndrome (trisomy 21) is the single most common chromosomal cause of intellectual disability, occurring in approximately 1 in 700–1000 live births in India. It is also the leading indication for genetic counselling in paediatric practice. Final-year students and interns are expected to recognise the syndrome clinically, interpret a standard karyotype, communicate the diagnosis empathetically to families, and explain the recurrence risk — which varies significantly depending on the cytogenetic mechanism. Errors in counselling (for instance, quoting a blanket 1% recurrence when the cause is a balanced translocation) can mislead families about their reproductive choices. Understanding this condition also connects to broader topics in neonatology, cardiology, and developmental medicine.
RECALL
Before proceeding, recall the following foundational concepts:
• Chromosomes: Humans have 46 chromosomes (23 pairs). Non-disjunction is the failure of chromosome pairs to separate during meiosis, resulting in a gamete with an extra (n+1) or missing (n−1) chromosome. When an (n+1) gamete fuses with a normal gamete, the zygote has 47 chromosomes — an aneuploidy.
• Robertsonian translocation: A structural rearrangement in which two acrocentric chromosomes (chromosomes 13, 14, 15, 21, 22) fuse at their centromeres, forming a single large chromosome. The carrier has 45 chromosomes in number but all chromosomal material is retained.
• Karyotype: A display of an individual's chromosomes arranged by size and banding pattern. A normal karyotype is 46,XX (female) or 46,XY (male). Notation 47,XX,+21 denotes a female with three copies of chromosome 21.
• Recall from PE-foundation: major developmental milestones — gross motor, fine motor, language, and social — and how global developmental delay differs from isolated delay.
Clinical Presentation of Down Syndrome
Down syndrome presents with a distinctive combination of dysmorphic features, hypotonia, and developmental delay that is recognisable at birth in most cases. The clinical features are the direct consequence of gene-dosage imbalance from the extra copy of chromosome 21, which contains genes affecting facial morphogenesis, brain development, cardiac embryology, and immune regulation. The extra chromosome 21 encodes approximately 300 genes, and the dosage effect of even a subset of these genes is sufficient to produce the recognisable phenotype across multiple organ systems. Recognising the full constellation of features enables early diagnosis, guides the investigations needed, and provides the foundation for counselling. Clinicians must be able to identify both the cardinal features (which are highly suggestive in isolation) and the supporting features (which reinforce the clinical suspicion and help differentiate Down syndrome from other causes of neonatal hypotonia and dysmorphism). The diagnosis should always be confirmed by karyotype rather than clinical diagnosis alone, but recognising the phenotype is the essential first step that triggers investigation.
Facial and head features are the most prominent:
• Upslanting palpebral fissures (lateral canthus higher than medial) — pathognomonic orientation.
• Epicanthal folds — medial skin folds partially covering the inner corner of the eye.
• Brushfield spots — small, whitish speckling of the iris (visible in light-coloured irides).
• Flat nasal bridge and small, low-set ears.
• Protruding tongue (macroglossia relative to small oral cavity) and high-arched palate.
• Brachycephaly (flat occiput) and microcephaly.
Musculoskeletal and hand findings:
• Single palmar (simian) crease — a single transverse crease replacing the normal two creases (present in ~50% of cases; also seen in normals ~5%).
• Clinodactyly of fifth finger — inward curving of the little finger due to hypoplastic middle phalanx.
• Short, broad hands with sandal gap between the first and second toes.
Systemic features:
• Generalised hypotonia — present in nearly all neonates; contributes to feeding difficulties and delayed motor milestones.
• Intellectual disability — universal; typically mild to moderate (IQ 50–70), rarely severe. Language development is more delayed than motor development.
• Short stature — below 3rd centile for age on standard growth charts (use DS-specific charts for monitoring).
Developmental trajectory: Children with Down syndrome follow the normal developmental sequence but at a slower pace. Early intervention (physiotherapy, occupational therapy, speech therapy) started within the first year of life significantly improves outcomes. Inclusive education is now recommended by IAP.
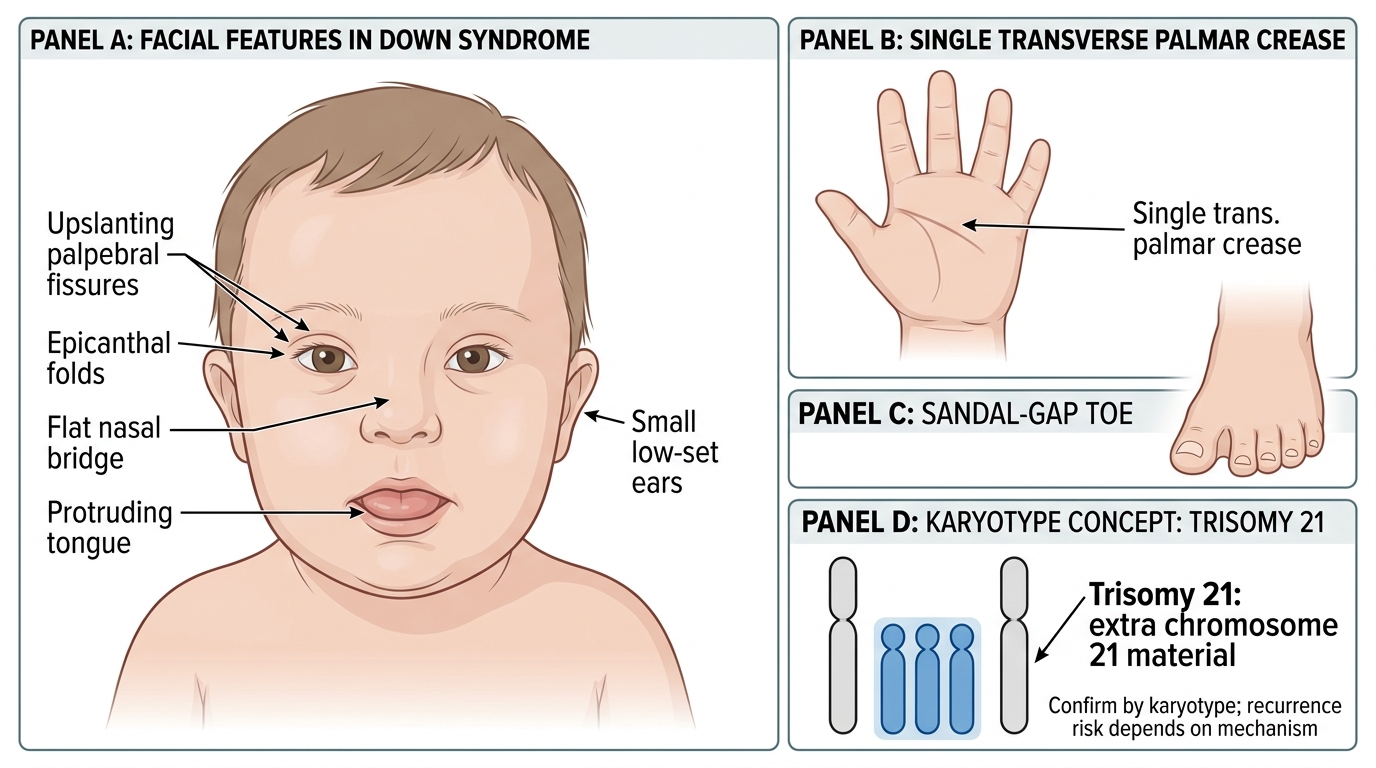
Characteristic Clinical Features of Down Syndrome
Genetic Basis and Pathophysiology
Down syndrome arises from three distinct cytogenetic mechanisms, each with a different recurrence risk. Understanding these mechanisms is essential not only for examination purposes but because it directly determines the counselling you give to the family. Grouping all Down syndrome into a single recurrence-risk category is one of the most clinically dangerous errors in genetics counselling.
1. Free trisomy 21 (~95% of cases)
This is the most common mechanism. It results from meiotic non-disjunction, usually occurring during maternal meiosis I (approximately 90% of free trisomies) or meiosis II. One gamete carries two copies of chromosome 21 instead of one; when it fuses with a normal gamete, the zygote has 47 chromosomes. The karyotype is written 47,XX,+21 (female) or 47,XY,+21 (male). The risk of non-disjunction increases sharply with advancing maternal age: the risk is ~1 in 1,250 at age 25, ~1 in 350 at age 35, and ~1 in 100 at age 40. Recurrence risk: approximately 1% above the background age-related risk (i.e., the empirical recurrence is ~1% + maternal-age-specific risk). Parental karyotypes are normal.
2. Robertsonian translocation (~4% of cases)
Here, the extra chromosome 21 material is attached to another acrocentric chromosome — most commonly chromosome 14, giving rise to a t(14;21) translocation, or rarely t(21;21) or t(21;22). The total chromosome count in the affected child is 46, but an extra copy of chromosome 21's long arm is present attached to chromosome 14. The karyotype is written 46,XX,der(14;21)(q10;q10). This type is crucial because it may be inherited from a carrier parent whose karyotype shows only 45 chromosomes (all material present, but rearranged). Recurrence risk by carrier status:
- Maternal carrier of t(14;21): ~10–15% risk of an affected offspring per pregnancy.
- Paternal carrier of t(14;21): ~2–5% risk per pregnancy (lower because of segregational disadvantage in sperm).
- t(21;21) translocation: 100% recurrence risk — every gamete carries the translocation, and every liveborn offspring will have Down syndrome.
Because this form may be familial, parental karyotyping is mandatory whenever a translocation is identified in the child.
3. Mosaicism (~1% of cases)
Mosaicism results from post-zygotic mitotic non-disjunction — the zygote starts out normal (46 chromosomes), but an error during early cell division creates a clone of cells with 47 chromosomes. The individual has two cell lines: 47,+21 and 46,normal. The phenotype is often milder (depending on the proportion of trisomic cells), and the intellectual disability may be less severe. Recurrence risk: empirical risk similar to free trisomy (~1% + maternal age); parental karyotypes are normal.
Karyotype interpretation: A karyogram displays chromosomes arranged in pairs by size and banding pattern. To identify trisomy 21, count the chromosomes in pair 21 — three copies are present instead of two. The rest of the pairs appear normal in free trisomy. Learners should be able to distinguish: (a) a normal 46,XX or 46,XY karyotype, (b) a 47,+21 free trisomy karyotype, and (c) a translocation karyotype where the total count is 46 but one chromosome 14 appears unusually large due to the attached 21.
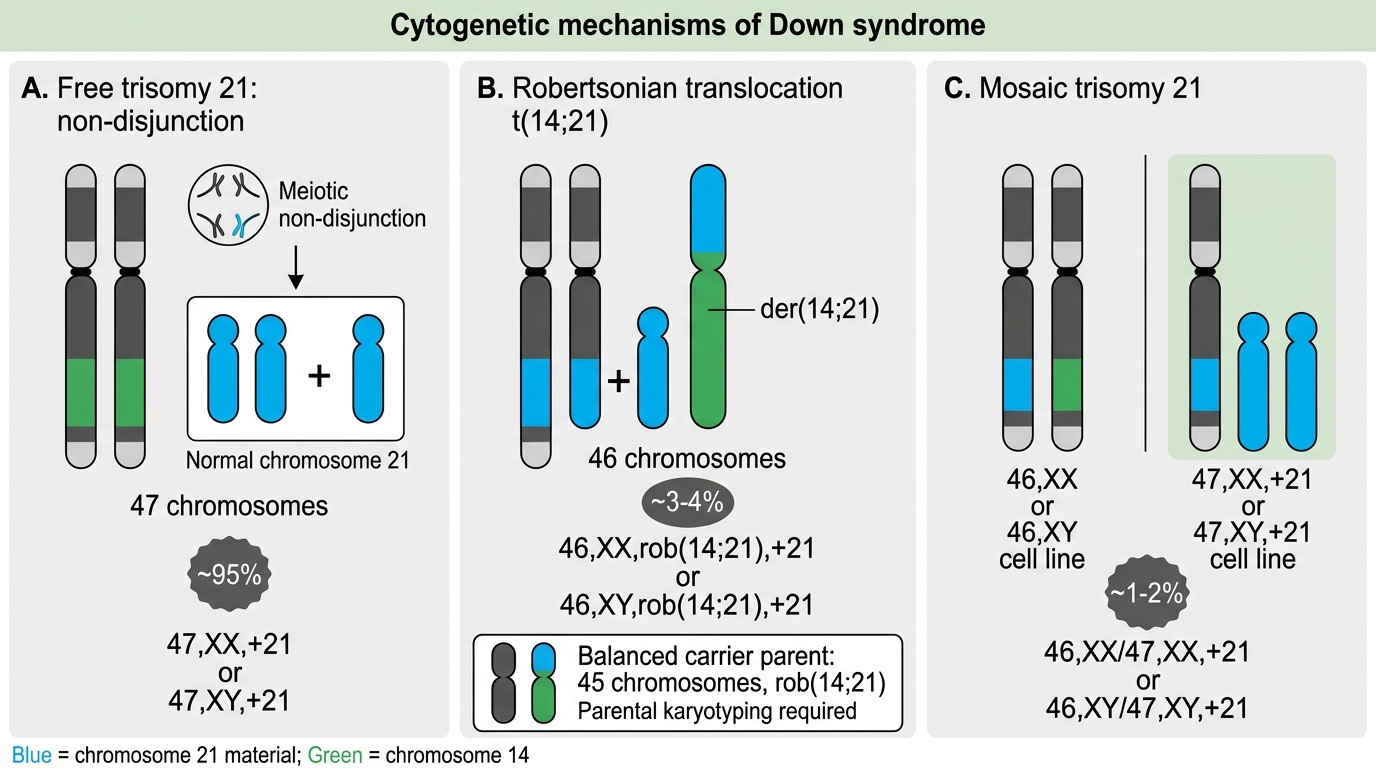
Cytogenetic Mechanisms of Down Syndrome
SELF-CHECK
A 3-month-old child with Down syndrome features has a karyotype showing 46 chromosomes, with one chromosome 14 appearing abnormally large. What is the most likely cytogenetic mechanism and what investigation should both parents undergo?
A. Free trisomy 21 — no further parental testing needed
B. Robertsonian translocation — both parents need karyotyping to identify a carrier
C. Mosaicism — peripheral blood karyotype of the mother only
D. Trisomy 21 mosaicism — skin biopsy karyotype of both parents
Reveal Answer
Answer: B. Robertsonian translocation — both parents need karyotyping to identify a carrier
A child with Down syndrome phenotype and 46 chromosomes on karyotype (with an abnormally large chromosome 14) indicates a robertsonian translocation, most likely t(14;21). The extra chromosome 21 material is attached to chromosome 14, giving a total count of 46. This form can be inherited from a carrier parent who has only 45 chromosomes but all chromosomal material. Both parents must be karyotyped: if either is a carrier of t(14;21), the recurrence risk is 10–15% (maternal) or 2–5% (paternal). If neither parent is a carrier, it is a de novo translocation and the recurrence risk is low.
Complications and Associated Conditions
Down syndrome is a multi-system disorder, and a significant proportion of morbidity and mortality stems from associated conditions rather than from the intellectual disability itself. Systematic surveillance for these complications is a core responsibility of the paediatrician caring for a child with Down syndrome. Understanding which complications are life-threatening (cardiac, haematological) and which are manageable with proactive screening (hypothyroidism, hearing loss, atlantoaxial instability) allows the clinician to prioritise workup appropriately. The chromosome 21 gene-dosage effect disrupts organogenesis in multiple systems — the endocardial cushion morphogenesis pathway (AVSD), intestinal rotation and atresia (duodenal atresia, Hirschsprung disease), immune regulation and cell cycle control (leukaemia), collagen synthesis (atlantoaxial ligament laxity), and thyroid function. Recognising this multi-system biology prevents the error of treating Down syndrome only as an intellectual disability condition and missing treatable medical co-morbidities. The organ-system approach below reflects the surveillance schedule endorsed by the American Academy of Pediatrics (2022) and adapted by IAP.
Cardiovascular (most common life-threatening complication):
• Congenital heart disease is present in approximately 40–50% of children with Down syndrome.
• Atrioventricular septal defect (AVSD) — the most characteristic and common lesion (~40% of CHD in Down syndrome); results from failure of fusion of the endocardial cushions during cardiac morphogenesis. AVSD causes a large left-to-right shunt, pulmonary hypertension, and cyanosis if untreated.
• Other common lesions: ventricular septal defect (VSD), atrial septal defect (ASD), tetralogy of Fallot, and patent ductus arteriosus (PDA).
• All children with Down syndrome require echocardiography in the neonatal period, regardless of clinical examination findings, because CHD may be silent initially.
Gastrointestinal:
• Duodenal atresia ("double bubble" sign on X-ray) — Down syndrome is the most common chromosomal association; presents with bilious vomiting in the neonate.
• Hirschsprung's disease — aganglionosis of the colon; presents with constipation and abdominal distension.
• Tracheo-oesophageal fistula and anorectal malformations — less common but recognised associations.
Orthopaedic:
• Atlantoaxial instability (AAI) — laxity of the transverse atlantal ligament (due to collagen abnormalities from chromosome 21 gene-dosage effects) can lead to subluxation of the C1–C2 joint. Prevalence ~15%; symptomatic instability <2%. Presents with neck pain, torticollis, or neurological signs (upper motor neuron signs, gait abnormality). Neck X-ray (flexion-extension lateral views): atlanto-dens interval >5 mm is abnormal.
Endocrine:
• Hypothyroidism — both congenital hypothyroidism (detectable on newborn screening) and autoimmune (Hashimoto's) thyroiditis are increased. Annual thyroid function testing (TSH) is recommended from birth onwards.
Haematological:
• Transient myeloproliferative disorder (TMD) — occurs in ~10% of neonates with Down syndrome; proliferation of blast cells that typically resolves spontaneously within 3 months. A minority (~20% of TMD) progress to acute myeloid leukaemia (AMKL).
• Acute lymphoblastic leukaemia (ALL) — risk is ~20-fold higher than in the general paediatric population.
Sensory:
• Hearing loss — present in ~75% (predominantly conductive due to recurrent otitis media, but sensorineural component also seen). Annual audiological assessment from 6 months of age.
• Visual defects — refractive errors, strabismus, and nystagmus; annual ophthalmology assessment.
Neurological:
• Seizure disorders in ~5–10%.
• Early-onset Alzheimer's disease — nearly universal by the 5th decade, due to the APP gene on chromosome 21 (amyloid precursor protein).
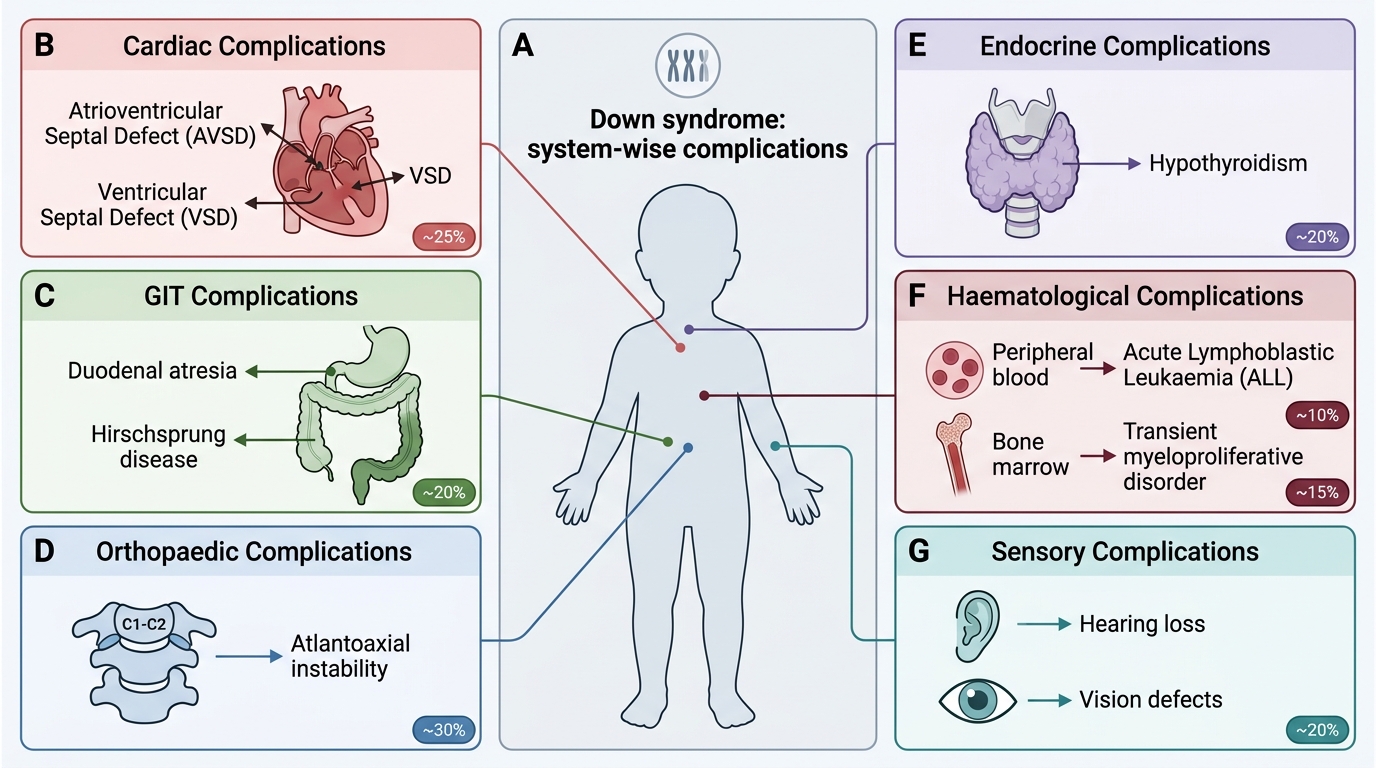
System-wise Complications of Down Syndrome