Page 2 of 14
PE29.1-3 | Down Syndrome — SDL Guide (Part 2)
Diagnosis and Investigation
The diagnosis of Down syndrome involves both prenatal screening/diagnosis (for detection before birth) and postnatal confirmation by karyotyping. The two are complementary strategies that serve different clinical purposes. Prenatal testing allows families to make informed reproductive choices; it encompasses a spectrum from low-risk non-invasive screening (which estimates the probability of trisomy 21) to definitive invasive diagnostic procedures (which yield a fetal karyotype). Postnatal evaluation, once clinical features prompt suspicion, focuses on two goals: confirming the cytogenetic diagnosis by karyotype and identifying associated conditions that require immediate management. It is important to distinguish screening from diagnosis in counselling — a high-risk screening result is not a diagnosis and must not be communicated as such. Equally, in the postnatal period, a clinical diagnosis of Down syndrome based on phenotype must always be confirmed by karyotype because the cytogenetic type (free trisomy vs translocation vs mosaicism) is unknown until the karyotype is available, and this directly affects recurrence-risk counselling.
Prenatal Screening (non-diagnostic — stratifies risk, does not confirm):
First-trimester combined screening (11–13+6 weeks):
• Nuchal translucency (NT) ultrasonography — measurement of fluid at the back of the fetal neck. NT >3.5 mm is associated with chromosomal anomalies including trisomy 21 and cardiac defects.
• Serum biochemistry: low PAPP-A (pregnancy-associated plasma protein A) and elevated free β-hCG.
• Combined detection rate: ~85–90%, false-positive rate ~5%.
Second-trimester quadruple (Quad) screen (15–20 weeks):
• Measures AFP (low), β-hCG (elevated), estriol (low), and inhibin-A (elevated).
• Detection rate ~75–80%.
Cell-free DNA (cfDNA) / Non-invasive Prenatal Testing (NIPT):
• Analyses fetal DNA in maternal blood from ~10 weeks gestation.
• Detection rate >99% for trisomy 21; low false-positive rate.
• A screening test, not diagnostic — a positive result requires confirmation by invasive testing.
Prenatal Diagnosis (definitive — provides karyotype):
• Chorionic villus sampling (CVS): At 10–13 weeks; aspirates placental tissue; allows early diagnosis. Procedure-related miscarriage risk ~0.5–1%.
• Amniocentesis: At 15–20 weeks; aspirates amniotic fluid containing fetal cells; miscarriage risk ~0.5%.
• Both yield a full fetal karyotype by conventional cytogenetics (G-banding) or FISH (faster, semi-quantitative for chromosomes 13, 18, 21, X, Y).
Postnatal diagnosis and investigation panel:
• Karyotype — confirmatory; identifies cytogenetic type (free trisomy vs translocation vs mosaicism); mandatory for counselling.
• Echocardiography — detect CHD in all neonates.
• Thyroid function (TSH + free T4) — congenital hypothyroidism screening (already included in national newborn screening in some states; check and confirm).
• Complete blood count — screen for TMD (blasts in neonatal period), polycythaemia.
• Hearing screen (BERA/OAE) — before 1 month.
• Ophthalmology assessment — red reflex, strabismus by 6 months.
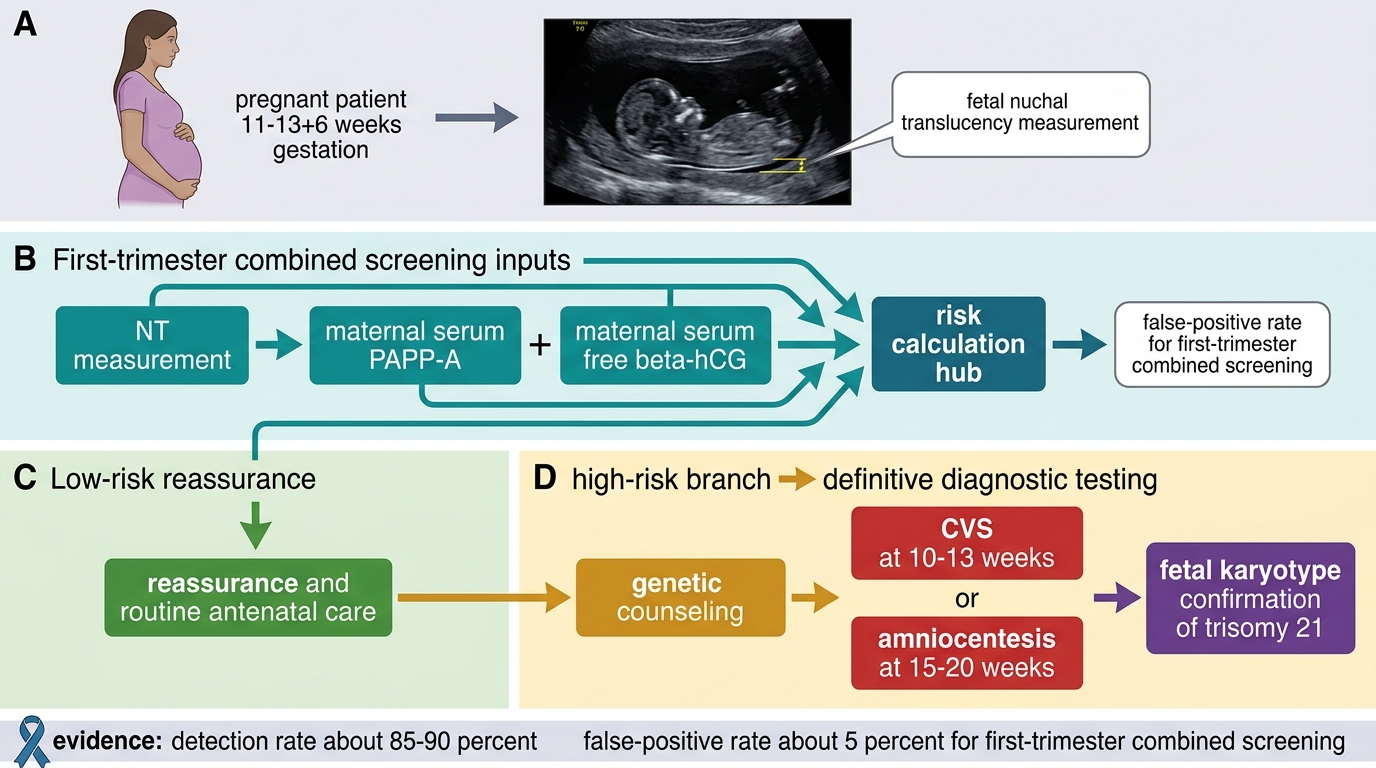
Prenatal Screening and Diagnosis Pathway for Down Syndrome
SELF-CHECK
A pregnant woman at 12 weeks gestation has first-trimester combined screening showing NT 4.2 mm and low PAPP-A. The result is reported as high risk for trisomy 21. What is the most appropriate next step?
A. Repeat NT at 16 weeks to confirm
B. Perform amniocentesis for karyotype confirmation
C. Offer cfDNA (NIPT) as the next diagnostic step
D. Offer CVS for definitive karyotype diagnosis
Reveal Answer
Answer: D. Offer CVS for definitive karyotype diagnosis
A high-risk first-trimester combined screen is a strong indication for definitive prenatal diagnosis. CVS (chorionic villus sampling) at 10–13 weeks is the most appropriate next step at 12 weeks gestation, as it provides a full fetal karyotype, identifying the cytogenetic type (free trisomy vs translocation), and allows early decision-making. Amniocentesis is an alternative but performed later (15–20 weeks). NIPT (cfDNA) is another screening test, not diagnostic — a positive NIPT still requires confirmation by CVS or amniocentesis. Repeating NT is not a diagnostic step.
Management and Multidisciplinary Care
Management of Down syndrome is multidisciplinary and lifelong. It spans the neonatal period (identification and treatment of acute complications), early childhood (early intervention, surveillance), school age (inclusive education, behavioural support), and adulthood (monitoring for Alzheimer's disease, psychiatric disorders). The paediatrician acts as the coordinator of this care network, ensuring that the child receives timely specialist input from cardiology, surgery, audiology, ophthalmology, physiotherapy, speech therapy, and developmental paediatrics, while the family receives ongoing psychosocial support. The overarching goal has shifted dramatically in recent decades — from custodial care to active inclusion and supported independence. Evidence demonstrates that early intervention begun in infancy, inclusive schooling, and proactive medical surveillance together produce substantially better cognitive and social outcomes than deferred or segregated care. The paediatrician must be familiar with the IAP surveillance schedule so that no treatable complication is missed during the critical early years.
Neonatal period:
• Confirm diagnosis with karyotype; communicate empathetically with the family (see counselling section).
• Echocardiography: AVSD and other CHD require early cardiology referral. Surgical correction is typically undertaken in the first 3–6 months of life before pulmonary hypertension becomes irreversible.
• Evaluate for duodenal atresia if bilious vomiting — surgical repair (Kimura diamond-shaped duodenoduo denostomy).
• Feed support: hypotonia impairs suckling; occupational therapy input for feeding techniques.
Early intervention (0–3 years):
• Physiotherapy for hypotonia and gross motor delay.
• Occupational therapy for fine motor development and feeding.
• Speech and language therapy — critical because language delays are disproportionate.
• Early stimulation programmes (IAP recommends starting within the first month).
Surveillance schedule (IAP/AAP-adapted):
• Thyroid: TSH at birth, 6 months, 12 months, then annually.
• Hearing: BERA/OAE at birth, then audiologically every 6 months up to age 3, then annually.
• Vision: Ophthalmology at 6 months, then annually.
• Atlantoaxial instability: Neck X-ray (flexion/extension) at 3–5 years if asymptomatic; earlier if neurological symptoms. Restrict contact sports and trampolining if AAI confirmed on imaging.
• Dental: 6-monthly review from first tooth eruption (mouth-breathing and poor hygiene increase caries risk).
• Haematology: CBC in neonatal period for TMD; lower threshold for investigating fatigue or lymphadenopathy for leukaemia.
Education and psychosocial support:
• Inclusive education in mainstream schools with appropriate support is the current IAP recommendation.
• Behavioural issues: attention deficit, obsessive-compulsive behaviours, and autism spectrum features are more prevalent.
• Psychological support for parents through parent-support groups (Down Syndrome Federation of India).
Adult surveillance:
• From the 4th decade: annual cognitive assessment for Alzheimer's disease.
• Hypothyroidism, atlantoaxial instability, and cardiac disease continue to require surveillance.
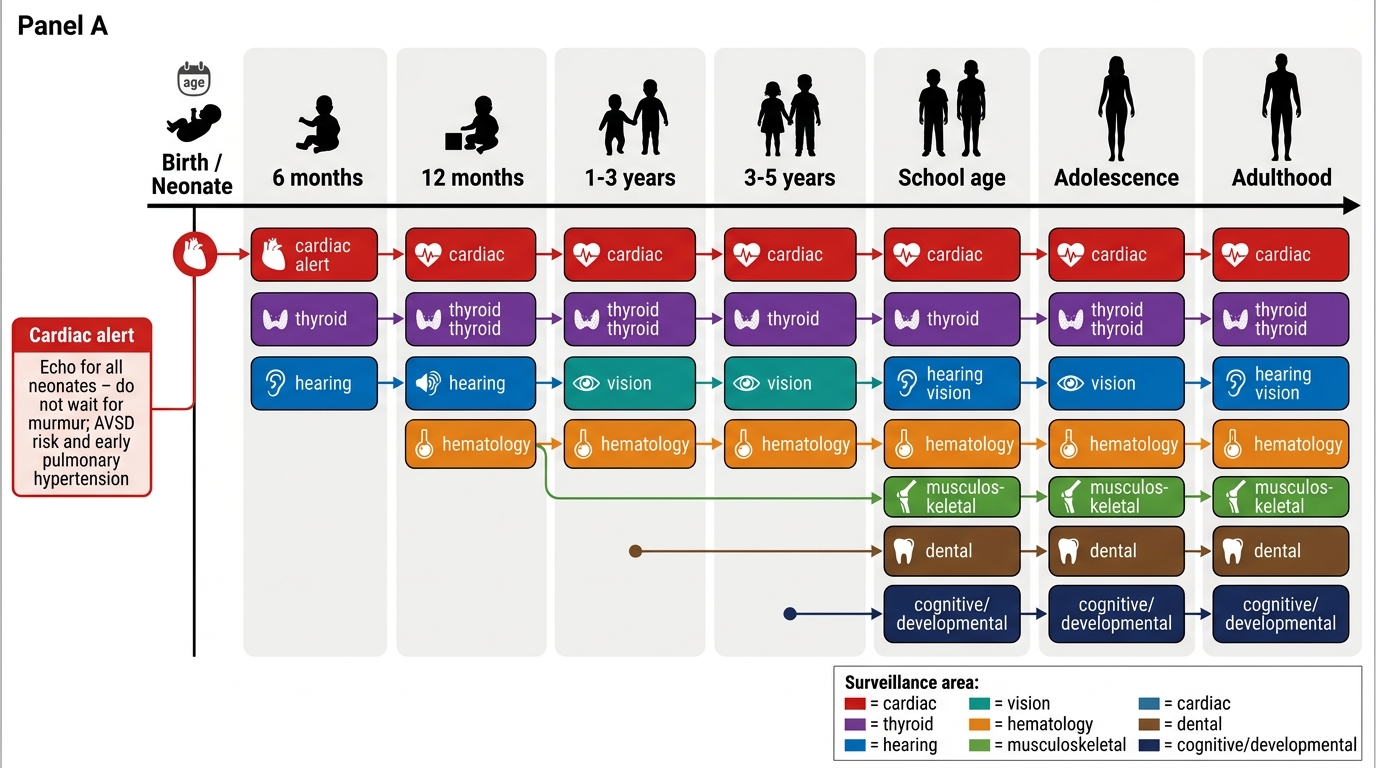
Down Syndrome Surveillance Timeline
CLINICAL PEARL
AVSD in Down syndrome — act before pulmonary hypertension sets in. Atrioventricular septal defect is the hallmark cardiac lesion of Down syndrome (~40% of CHD cases). Children with DS and AVSD develop pulmonary arterial hypertension faster than chromosomally normal children with the same defect — often by 6 months of age. If surgical correction is delayed beyond the development of irreversible pulmonary vascular disease (Eisenmenger syndrome), the child becomes inoperable. Routine echocardiography in all neonates with Down syndrome (even with a clinically normal examination) is therefore mandatory, not optional. Never defer cardiac workup waiting for an obvious murmur — many large AVSDs are clinically silent early.
Genetic Counselling — Present Child and Future Pregnancies
Genetic counselling in Down syndrome addresses two distinct clinical scenarios, each requiring a different conversation: (1) supporting the family in caring for the child who already has Down syndrome, and (2) advising the family on recurrence risk in future pregnancies. These two conversations differ in their purpose, timing, and content, and conflating them is a recognised communication error. The immediate post-diagnosis conversation should focus on the present child — the diagnosis, what it means for the child's development, what investigations are needed, what treatments are available, and what the realistic prognosis looks like with appropriate support. Recurrence risk information, while important, should generally be deferred until the family has had time to emotionally process the diagnosis, unless they explicitly ask for it. Furthermore, no recurrence-risk figure should ever be quoted before the karyotype result is available, because the recurrence risk differs dramatically between cytogenetic types. The counsellor must be genuinely non-directive — this means presenting information completely and accurately while respecting the family's autonomy in all reproductive decisions.
Provided image
Counselling for the present child:
The immediate post-diagnosis conversation should be empathic, unhurried, and non-directive. Key elements:
• Deliver the news in a private setting with both parents present if possible.
• Use clear, non-stigmatising language: say "Down syndrome" rather than "Down's baby" or "mongolism" (the latter is obsolete and offensive).
• Explain what Down syndrome means in terms of development (intellectual disability, motor delay), health surveillance needs, and realistic prognosis (many adults with DS lead semi-independent lives with appropriate support).
• Emphasise early-intervention services and parent-support resources.
• Avoid a negative prognosis frame — outcomes have improved dramatically with intervention and inclusion.
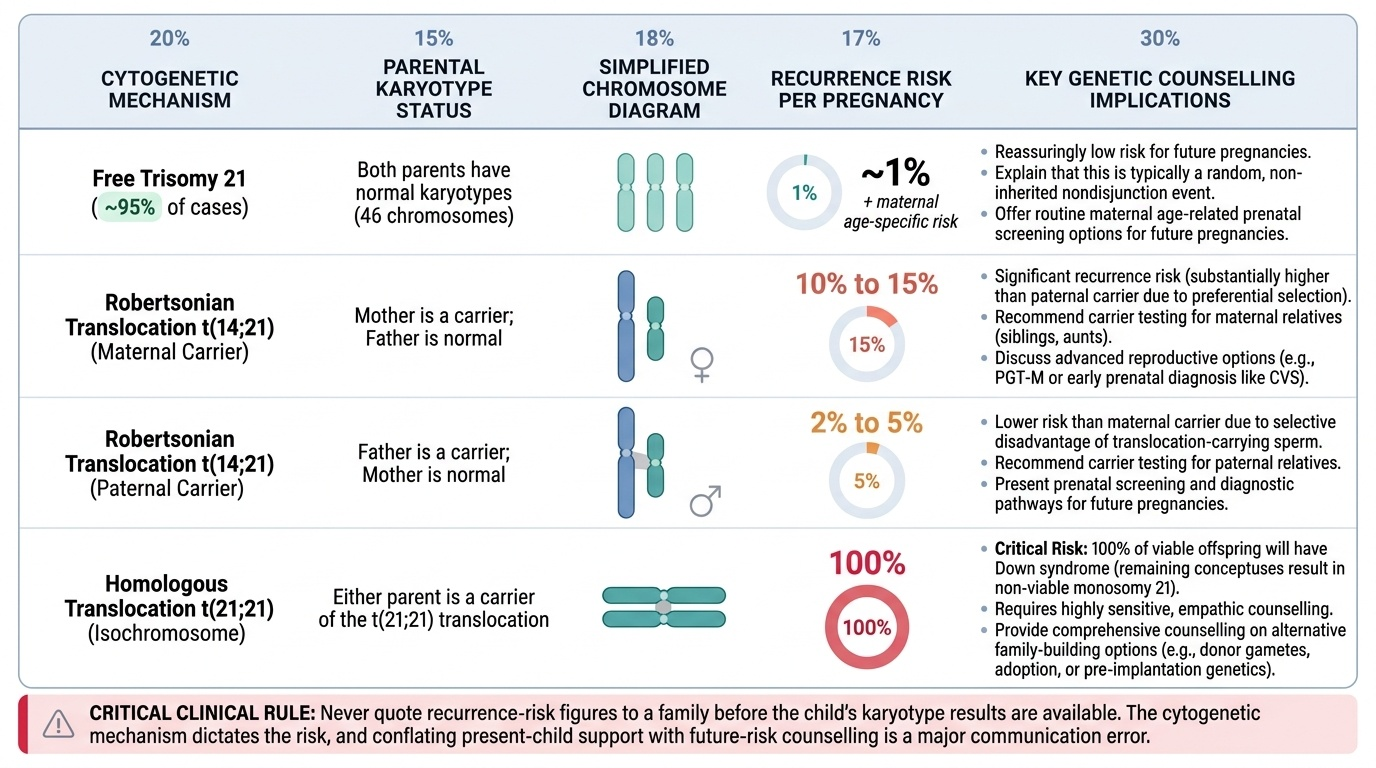
Recurrence-risk counselling — mechanism determines risk:
Before quoting any recurrence risk, the cytogenetic type MUST be known (karyotype of the child, and if translocation, karyotype of both parents).
| Cytogenetic type | Parental karyotype | Recurrence risk per pregnancy |
|---|---|---|
| Free trisomy 21 (~95%) | Both normal | ~1% + maternal age-specific risk |
| Translocation t(14;21) — maternal carrier | Mother: 45 chromosomes (carrier) | ~10–15% |
| Translocation t(14;21) — paternal carrier | Father: 45 chromosomes (carrier) | ~2–5% |
| Translocation t(14;21) — de novo in child | Both parents: normal 46 | ~1% (low) |
| Translocation t(21;21) | Either parent carries t(21;21) | 100% — all conceptuses affected |
| Mosaicism (~1%) | Both normal | ~1% + maternal age-specific risk |
Key counselling points for recurrence:
• For free trisomy: reassure that the recurrence risk is low (~1% above age risk); no parental karyotyping is routinely needed unless maternal age is young (<30 years without other risk factors).
• For translocation: mandatory parental karyotyping before quoting any risk. If either parent is a carrier, extended family members should also be offered karyotyping (familial cascade testing).
• All parents should be offered prenatal testing in future pregnancies, regardless of type.
• Acknowledge the emotional weight of the question; do not rush the recurrence-risk discussion in the same breath as the diagnosis.
SELF-CHECK
Parents of a child with Down syndrome due to free trisomy 21 (47,XX,+21) ask you the recurrence risk for their next pregnancy. The mother is 28 years old. Parental karyotypes are both normal. What is the most accurate counselling?
A. The recurrence risk is 25%, similar to autosomal recessive conditions
B. The recurrence risk is approximately 1% above the maternal-age-specific background risk
C. The recurrence risk is 10–15% because translocation carriers have higher risk
D. The recurrence risk is zero because both parents are chromosomally normal
Reveal Answer
Answer: B. The recurrence risk is approximately 1% above the maternal-age-specific background risk
Free trisomy 21 results from meiotic non-disjunction in a parent with a normal karyotype. The empirical recurrence risk is approximately 1% above the background maternal-age-specific risk. For a 28-year-old, the baseline age-related risk is already low (~1 in 1,000), so the total recurrence risk is approximately 1–2%. This is importantly different from translocation Down syndrome, where a carrier parent carries a 10–15% (maternal) or 2–5% (paternal) recurrence risk. A recurrence risk of 25% would apply to autosomal recessive conditions, which require homozygous loss-of-function mutations — not applicable here. Zero risk is also incorrect; while the risk is low, it is not zero.
Self-Assessment
Use these questions to consolidate your understanding of Down syndrome before you assess a child or counsel a family in the clinical setting. Self-assessment at this stage is not merely about recalling facts — it is about testing whether you can integrate genetic, clinical, investigative, and counselling knowledge into a coherent clinical response. The questions below are structured to mirror the complexity of real OSCE stations and long-case discussions: they require you to explain mechanisms, apply recurrence-risk knowledge to specific scenarios, and articulate counselling strategies that are both accurate and empathic. A student who can answer all six questions below without prompting has achieved the level of competency required by PE29.1, PE29.2, and PE29.3. Ensure you can answer each without referring back to the text.
Structured self-check questions:
- Name the three cytogenetic types of Down syndrome. For each, state the approximate frequency, the underlying mechanism, the karyotype notation, and the parental karyotype finding.
- A 2-day-old neonate has Down syndrome features. Echocardiography shows a large atrioventricular septal defect. The parents want to know why surgery is needed urgently. Explain the pathophysiology and the consequence of delayed surgery.
- How would you interpret the following karyotype report: "46,XX,der(14;21)(q10;q10)"? What further investigation is mandatory and why?
- A couple with a t(21;21) carrier mother asks about future pregnancies. What recurrence risk do you quote and what options do you offer?
- Enumerate five system-wise complications of Down syndrome that require proactive surveillance, and state the recommended screening test and age for each.
- How does the counselling for the family of a newly diagnosed child differ from the counselling for recurrence risk? What are the key principles of breaking a chromosomal diagnosis to parents?
Clinical skills integration: Practice reading a karyogram provided by your clinical genetics tutor. Identify: (a) the total chromosome count, (b) sex chromosome pair, (c) whether chromosome 21 has two or three copies, (d) whether any chromosome shows an unusual size suggesting a translocation.
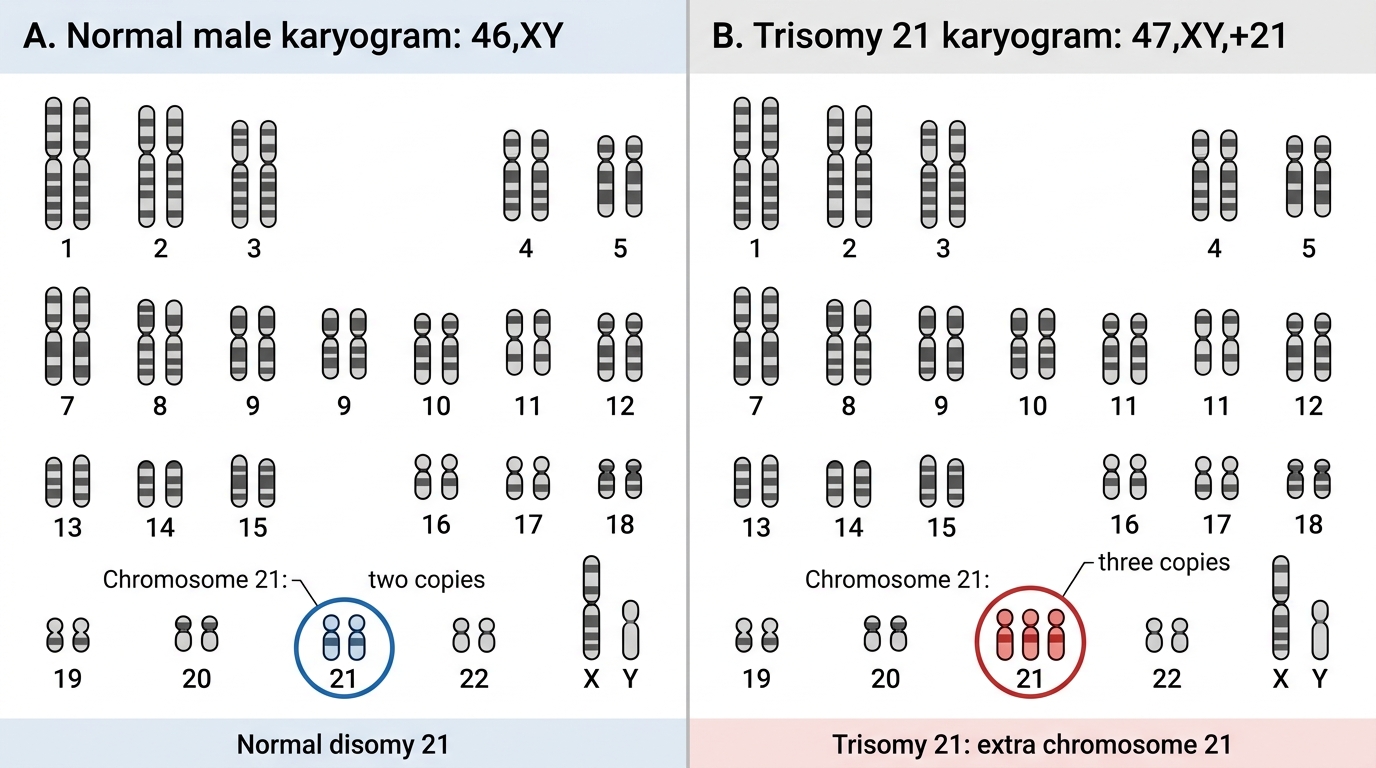
Normal 46,XY vs Trisomy 21 Karyogram