Page 7 of 14
PE29.5 | Klinefelter Syndrome — SDL Guide
Learning Objectives
- Describe the genetic basis of Klinefelter syndrome including the 47,XXY karyotype, mechanism of non-disjunction, and mosaic/higher-grade variants
- Identify the characteristic clinical features of Klinefelter syndrome in childhood, at puberty, and in adulthood
- Explain the pathophysiology of primary hypogonadism, azoospermia, and gynaecomastia in Klinefelter syndrome
- Outline the investigation pathway including karyotype, gonadotropin profile, and fertility assessment
- Describe the management plan including testosterone replacement, gynaecomastia management, and fertility counselling via TESE and ICSI
- Counsel patients and families regarding recurrence risk and reproductive options
INSTRUCTIONS
Klinefelter syndrome (47,XXY) is the most common sex-chromosome aneuploidy in males and the most common cause of male hypogonadism and non-obstructive azoospermia. With a birth prevalence of approximately 1 in 500–600 live male births, it is encountered in paediatric, endocrine, and urology clinics. Despite this prevalence, the condition is significantly under-diagnosed — the majority of affected males are not diagnosed until adulthood when they present with infertility or androgen deficiency. Final-year students must understand the cytogenetic basis, recognise the clinical features at each life stage, interpret the hormonal profile, and counsel appropriately about testosterone replacement and fertility options.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 22 — Chromosomal Disorders (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 588 — Klinefelter Syndrome (textbook)
- Groth KA et al. — Klinefelter syndrome: a clinical update (2013), Journal of Clinical Endocrinology & Metabolism (guideline)
- Indian Academy of Pediatrics — Guidelines on Developmental Disorders (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 16-year-old boy is brought to the paediatric outpatient clinic because he has not developed a beard, his voice has not broken, and his classmates comment that his chest appears 'like a girl's'. His height is 183 cm (tall for his family). On examination, both testes are palpable but measure approximately 2 mL bilaterally and feel unusually firm. He has mild bilateral gynaecomastia and sparse pubic hair. His mother asks: 'Is this just late puberty?' You suspect a chromosomal condition. What is the most likely diagnosis and the single most important investigation?
WHY THIS MATTERS
Klinefelter syndrome (47,XXY) is the most common sex-chromosome aneuploidy in males, affecting approximately 1 in 500–600 live male births. It is the single most common genetic cause of male infertility, accounting for approximately 10% of all azoospermic men. Despite this prevalence, the majority of affected males remain undiagnosed or are diagnosed only in adulthood when they present for infertility evaluation. This diagnostic gap matters clinically because earlier diagnosis enables timely testosterone replacement, educational support for language and learning difficulties, and psychological preparation for fertility challenges. Paediatricians play a pivotal role in recognising the condition during childhood or early puberty, before the diagnostic opportunity is lost to attrition.
RECALL
Before proceeding, recall the following foundational concepts:
• Sex chromosomes and karyotype: Normal male karyotype = 46,XY. The presence of a Y chromosome determines male sex differentiation; the X chromosome carries approximately 800 genes. In 47,XXY, the extra X chromosome is present in addition to the normal XY.
• Normal male puberty: First sign is testicular enlargement (Tanner stage G2), beginning age 9–14 years. Testicular volume reaches 15–25 mL by adulthood. FSH stimulates seminiferous tubules (spermatogenesis); LH stimulates Leydig cells (testosterone production).
• Gonadotropin regulation: Testosterone and inhibin B provide negative feedback on FSH/LH. In primary gonadal failure, testosterone is low → feedback lost → FSH and LH rise = hypergonadotropic hypogonadism. In secondary (hypothalamic/pituitary) failure, FSH and LH are both low.
• Azoospermia: Complete absence of sperm in ejaculate; differentiated into obstructive (normal spermatogenesis, blocked ducts) and non-obstructive (defective spermatogenesis — as in Klinefelter).
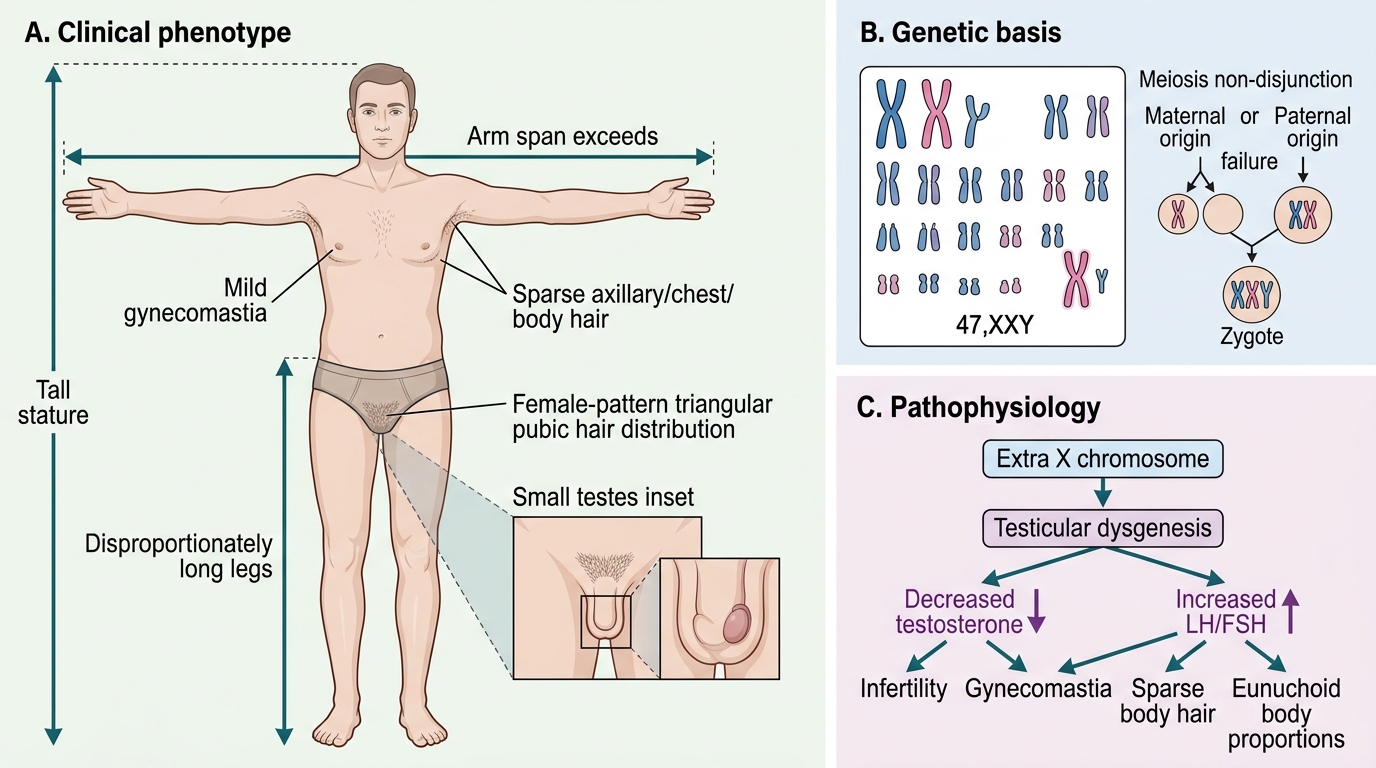
Clinical Presentation of Klinefelter Syndrome
Klinefelter syndrome is characterised by a remarkably variable phenotype — many affected males have mild or non-specific features, which explains the high rate of under-diagnosis. The classic full 47,XXY phenotype is most recognisable at puberty, when the contrast between tall stature with long limbs and small testes with incomplete virilisation becomes apparent. However, the condition has features across all life stages, and a paediatrician who knows what to look for can make the diagnosis in infancy or childhood rather than waiting for the pubertal presentation. The key insight is that the testes are normal-appearing in infancy and childhood, so the diagnosis in pre-pubertal boys relies on the softer clues of language delay, tall stature relative to the family, and learning difficulties. Only at puberty do the testes fail to enlarge appropriately, providing the cardinal physical finding that makes the diagnosis obvious if the examiner is looking for it.
Infancy and childhood (pre-pubertal):
• Language delay — the most consistent early finding; approximately 70–80% of boys with Klinefelter have delayed language development, reading difficulties, or learning disabilities. This is the most frequently missed early clue.
• Mild hypotonia and motor delay in infancy (less common than in Down syndrome).
• Tall stature relative to mid-parental height; long legs relative to trunk (eunuchoid proportions — arm span > height, lower segment > upper segment).
• Small testes may be noted on routine examination, though normal testicular size in childhood means this may not be apparent until puberty.
Puberty and adolescence (the classic presentation):
• Incomplete virilisation: Puberty begins at a normal age but fails to progress fully. Voice change is delayed or incomplete; beard and body hair remain sparse.
• Small, firm testes — the cardinal physical finding. Both testes are typically <4 mL bilaterally in post-pubertal males with Klinefelter (normal adult volume: 15–25 mL). The firmness reflects fibrosis and hyalinisation of the seminiferous tubules.
• Gynaecomastia — bilateral, usually non-tender breast tissue enlargement, present in approximately 50–75% of post-pubertal males. Results from an increased oestradiol-to-testosterone ratio due to peripheral aromatisation of androgens to oestrogens.
• Tall stature: final height typically 5–10 cm above mid-parental height centile.
Adulthood:
• Infertility — the most common presenting complaint in undiagnosed adults; azoospermia is nearly universal in 47,XXY males due to seminiferous tubule failure.
• Androgen deficiency symptoms: Reduced libido, erectile dysfunction, fatigue, decreased muscle mass, osteoporosis.
• Metabolic syndrome, type 2 diabetes, and cardiovascular risk are increased.
Klinefelter Syndrome: Clinical Features and Genetic Basis
Genetic Basis and Pathophysiology
Klinefelter syndrome results from the presence of an extra X chromosome in a phenotypic male, giving the karyotype 47,XXY. The extra X is maternally derived in approximately 50% of cases and paternally derived in approximately 50% — unlike trisomy 21, where the extra chromosome is predominantly of maternal origin. The mechanism is meiotic non-disjunction during either maternal or paternal gametogenesis. Unlike Down syndrome, the risk of Klinefelter syndrome does not increase significantly with maternal age, though a slight maternal age effect exists for the maternally-derived cases. The extra X chromosome is variably inactivated in different tissues, which contributes to the phenotypic variability.
Cytogenetic variants:
1. Classic 47,XXY (~80% of Klinefelter syndrome)
The karyotype shows 47 chromosomes with two X chromosomes and one Y. This is the most common variant and produces the full clinical spectrum described above. The extra X chromosome is largely inactivated (X-inactivation/Lyon hypothesis), but genes in the pseudoautosomal regions and other X-escaping genes have double dosage, contributing to the phenotype.
2. Mosaicism 46,XY/47,XXY (~10–20% of cases)
Post-zygotic non-disjunction produces two cell lines. The 46,XY cell line may partially rescue testicular function — mosaic Klinefelter may have less severe features, larger testes, and occasionally some residual spermatogenesis.
3. Higher-grade aneuploidies (rare): 48,XXXY; 49,XXXXY
With each additional X chromosome, the phenotype is more severe: greater intellectual disability, more pronounced skeletal abnormalities, and more complete gonadal failure.
Pathophysiology of key features:
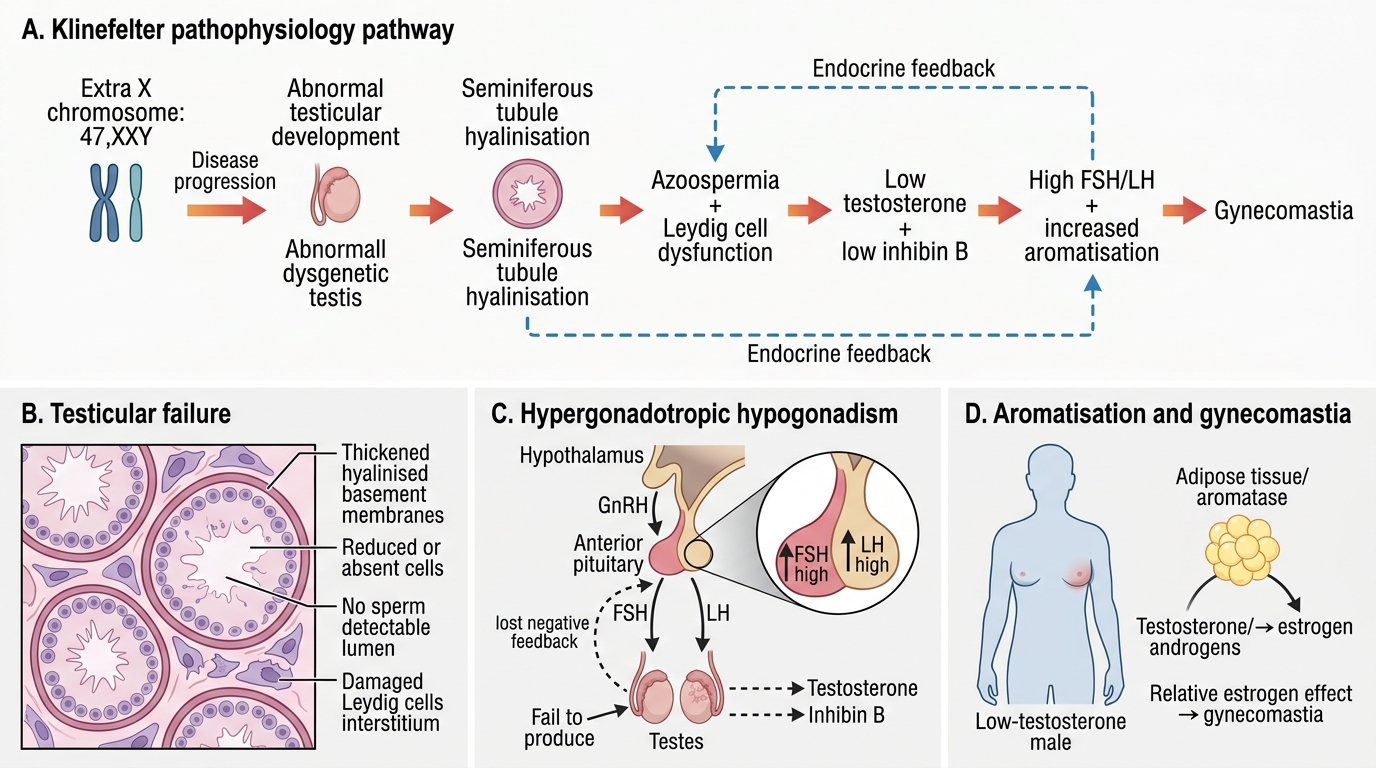
• Testicular failure and azoospermia: The extra X chromosome disrupts seminiferous tubule development during fetal life. By puberty, hyalinisation and fibrosis of the seminiferous tubules are nearly complete in classic 47,XXY, resulting in non-obstructive azoospermia. Leydig cells (which produce testosterone) are less severely affected but still fail to maintain normal testosterone levels by late puberty.
• Testosterone deficiency → incomplete virilisation: The combination of Leydig cell dysfunction and reduced Sertoli cell inhibin B output results in low testosterone and markedly elevated FSH and LH (hypergonadotropic hypogonadism).
• Gynaecomastia: The relative testosterone deficiency leads to a shift in the oestradiol-to-testosterone ratio. Peripheral aromatisation (by adipose tissue) converts androgens to oestrogens; with less testosterone substrate, the relative excess of oestrogen stimulates breast glandular tissue.
• Tall stature: The extra X chromosome contains genes (possibly PAR1 SHOX) that promote long-bone growth; combined with delayed epiphyseal closure from androgen deficiency, final height is increased.
Pathophysiology of Klinefelter Syndrome
SELF-CHECK
A 17-year-old boy with Klinefelter syndrome (47,XXY) has markedly elevated FSH and LH, low-normal testosterone, and azoospermia. What is the mechanism of the elevated gonadotropins, and what does this tell you about the level of the problem (hypothalamic-pituitary axis vs gonads)?
A. Elevated FSH/LH indicates a hypothalamic problem causing excess GnRH release
B. Elevated FSH/LH with low testosterone indicates primary gonadal failure (hypergonadotropic hypogonadism)
C. Elevated FSH/LH indicates a pituitary adenoma secreting excess gonadotropins
D. Elevated FSH/LH with low testosterone indicates secondary hypogonadism from pituitary failure
Reveal Answer
Answer: B. Elevated FSH/LH with low testosterone indicates primary gonadal failure (hypergonadotropic hypogonadism)
In Klinefelter syndrome, the testes are the site of failure — the seminiferous tubules and Leydig cells are damaged by the extra X chromosome. With low testosterone and no inhibin B from Sertoli cells, the negative feedback on the pituitary is lost. The pituitary responds by secreting increasing amounts of FSH and LH to try to stimulate the non-responsive gonads. This pattern — elevated FSH/LH with low testosterone — is called hypergonadotropic hypogonadism and localises the problem to the gonads (primary failure), not the hypothalamus or pituitary (which would produce low FSH/LH). This distinction is critical: treatment is testosterone replacement (not GnRH or gonadotropin stimulation).
Diagnosis and Investigation
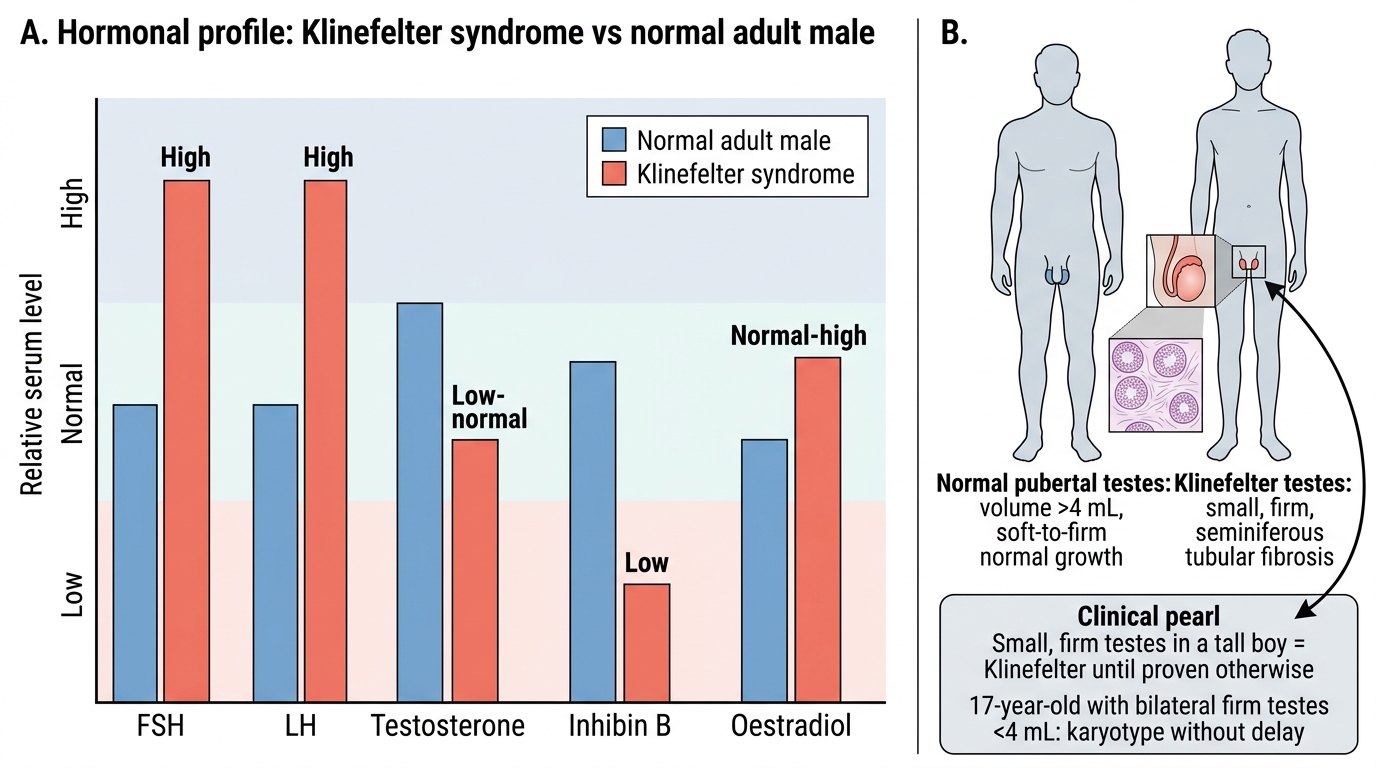
The diagnosis of Klinefelter syndrome is confirmed by karyotype. The clinical presentation — delayed or incomplete puberty, small firm testes, gynaecomastia, tall stature, or azoospermia in an adult — should trigger karyotype analysis. A hormonal profile provides supporting biochemical evidence and guides the urgency and specifics of management. Prenatal diagnosis is increasingly made by cfDNA screening. The investigation panel serves to confirm diagnosis, characterise the extent of gonadal failure, assess complications, and plan treatment. The hormonal pattern of elevated FSH and LH with low testosterone is the biochemical hallmark of primary gonadal failure in Klinefelter; it localises the defect to the gonads rather than the pituitary or hypothalamus and guides the choice of treatment (testosterone replacement, not gonadotropin stimulation). Understanding which investigation leads the diagnostic process and what each result means is essential for both the examination and for clinical practice.
Karyotype:
• Peripheral blood karyotype is confirmatory. For suspected mosaicism, a larger cell count (≥50 cells) should be analysed to avoid missing low-level mosaics.
• Identifies the specific variant: classic 47,XXY, mosaic 46,XY/47,XXY, or higher-grade aneuploidy.
• Prenatal diagnosis: cfDNA (NIPT) from ~10 weeks detects sex-chromosome aneuploidies; confirmation by amniocentesis provides definitive karyotype.
Hormonal profile:
• FSH: Markedly elevated (often >30–40 IU/L) — the most sensitive hormonal marker of seminiferous tubule failure; rises before testosterone falls.
• LH: Elevated due to Leydig cell failure.
• Testosterone: Low-normal in childhood; falls progressively through puberty; typically below the normal adult male range in untreated post-pubertal patients.
• Inhibin B: Very low or undetectable — produced by Sertoli cells; its absence reflects seminiferous tubule destruction.
• Oestradiol: Normal or mildly elevated (from peripheral aromatisation), contributing to gynaecomastia.
Semen analysis:
• In adolescents and adults: azoospermia (no sperm) is the rule in classic 47,XXY. Mosaic variants may have oligospermia.
Other investigations:
• Testicular ultrasound: Bilateral small testes with heterogeneous echotexture; calcifications may be present (reflecting tubular hyalinisation).
• Bone densitometry (DEXA): Osteoporosis risk from androgen deficiency.
• Thyroid function, fasting glucose, lipid profile: Metabolic surveillance.
• Neuropsychological assessment: Language, reading, and social cognition evaluation in children.
Hormonal Profile in Klinefelter Syndrome
CLINICAL PEARL
Small, firm testes in a tall boy = Klinefelter until proven otherwise. In normal puberty, testicular volume is the FIRST sign to increase (Tanner G2 = volume >4 mL). In Klinefelter syndrome, the testes remain small and firm despite apparent pubertal onset because the seminiferous tubules are being progressively replaced by fibrous tissue. The firmness is the distinguishing feature from other causes of small testes (e.g., cryptorchidism or hypothyroidism, where the testes are soft). A 17-year-old with bilateral firm testes of <4 mL should have a karyotype without delay — this is not 'constitutional delay of puberty', which would produce soft testes of normal size with delayed maturation.