Page 4 of 14
PE29.4 | Turner Syndrome — SDL Guide
Learning Objectives
- Describe the genetic basis of Turner syndrome including monosomy 45,X, mosaic variants, and structural X abnormalities
- Identify the characteristic somatic features and clinical presentations of Turner syndrome across the lifespan (neonate, child, adolescent)
- List the major associated complications: cardiovascular (bicuspid aortic valve, aortic coarctation), renal, and endocrine (hypothyroidism)
- Outline the investigation pathway including karyotype, gonadotropin profile, cardiac and renal imaging, and thyroid function
- Describe the management plan including growth hormone therapy, oestrogen replacement, cardiac surveillance, and fertility counselling
- Counsel parents and patients regarding recurrence risk and available reproductive options
INSTRUCTIONS
Turner syndrome is the most common sex-chromosome abnormality in phenotypic females, affecting approximately 1 in 2,000–2,500 live female births. It is a paradigm case for understanding how sex-chromosome gene dosage affects growth, puberty, cardiac development, and fertility. Final-year students must be able to recognise Turner syndrome clinically — in a neonatal girl with lymphoedema, in a child with unexplained short stature, or in an adolescent with primary amenorrhoea — and initiate the appropriate investigation pathway. Understanding the SHOX gene basis of short stature, the gonadal dysgenesis mechanism, and the critical importance of aortic surveillance are core competencies for the MBBS examination and clinical practice.
References
- Ghai Essential Pediatrics, 9th ed., Ch. 22 — Chromosomal Disorders (textbook)
- Nelson Textbook of Pediatrics, 21st ed., Ch. 589 — Turner Syndrome (textbook)
- Gravholt CH et al. — Clinical practice guidelines for the care of girls and women with Turner syndrome (2017), European Journal of Endocrinology (guideline)
- Indian Academy of Pediatrics — Growth Hormone Therapy in Children (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 14-year-old girl is brought by her parents because she has not yet started her periods and her height at 142 cm is well below her mid-parental height centile. She has always been shorter than her classmates but was otherwise healthy and performed adequately in school. On examination, she has a low posterior hairline, a short webbed neck, a broad chest with widely spaced nipples, and cubitus valgus. Her blood pressure in the right arm is 140/90 mmHg. You suspect a chromosomal syndrome. What is the most likely diagnosis, and what single most important investigation will you order first?
WHY THIS MATTERS
Turner syndrome is the most common sex-chromosome aneuploidy in females, with a birth prevalence of ~1 in 2,000–2,500 live female births. It accounts for approximately 10–15% of all spontaneous first-trimester abortions (since ~99% of 45,X conceptions are lethal in utero). In clinical practice, Turner syndrome is a diagnosis that paediatricians and general practitioners must not miss — because delayed diagnosis means missed growth hormone therapy during the critical window, undetected aortic coarctation causing hypertension and stroke risk, and unreplaced oestrogen causing osteoporosis. The condition spans the disciplines of endocrinology, cardiology, and reproductive medicine, making it a high-yield examination topic and a common long-case in MBBS finals.
RECALL
Before proceeding, recall the following foundational concepts:
• Sex chromosomes: Normal female karyotype = 46,XX; normal male = 46,XY. The X chromosome carries ~800 genes; the Y carries ~50. Females have two X chromosomes, but one is largely inactivated (Lyon hypothesis); the short arm pseudoautosomal region 1 (PAR1) escapes inactivation.
• Monosomy: Loss of one chromosome from a pair. Autosomal monosomies are universally lethal; sex-chromosome monosomy (45,X) is viable because most X-linked genes have the inactive copy anyway — but PAR1 genes (including SHOX) require two copies and are not inactivated.
• Normal puberty timeline: Thelarche ~10–11 years, pubarche ~10–11 years, menarche ~12–13 years. Primary amenorrhoea = no menarche by age 15 with secondary sex characteristics, or by age 13 without them.
• Gonadotropins: FSH and LH are secreted by the anterior pituitary; they stimulate gonadal oestrogen/testosterone production. In gonadal failure, the feedback loop is broken — FSH and LH rise (hypergonadotropic hypogonadism). In central causes, FSH/LH are low.
Clinical Presentation of Turner Syndrome
Turner syndrome presents differently depending on the age of detection — in the neonate, as lymphoedema and dysmorphic features; in childhood, as unexplained short stature; and in adolescence, as primary amenorrhoea with or without pubertal delay. The spectrum of somatic features varies significantly with the karyotype variant: full monosomy 45,X typically produces the classic phenotype, while mosaic Turner (45,X/46,XX) may have subtle or absent somatic features yet still require the same medical surveillance. A high index of suspicion is therefore essential — Turner syndrome must be considered in any girl with unexplained short stature regardless of whether she has dysmorphic features. In practice, the majority of Turner diagnoses are now made either prenatally (by cfDNA or amniocentesis) or in childhood during growth surveillance, rather than at the classic adolescent presentation with primary amenorrhoea. Recognising the full range of presentations prevents the diagnostic delay that deprives children of timely growth hormone therapy.
Neonatal presentation:
• Lymphoedema of the hands and feet — the earliest and most common neonatal feature; results from impaired lymphatic development. Pitting oedema of the dorsum of the hand and foot is often present at birth and resolves over the first few months.
• Webbed neck (pterygium colli) — redundant skin folds running from the mastoid to the acromion; caused by cystic hygroma that resolved in utero.
• Low posterior hairline — hairline extends lower on the neck than normal.
• Shield chest — broad, flat chest with widely spaced nipples; interpapillary distance >25% of chest circumference.
Childhood presentation:
• Short stature — the universal feature; follows a typical pattern of falling centiles from around age 2–3 years. By school age, height is typically 2–3 SD below the mean. The short stature is present even in mosaic Turner and even before gonadal failure produces oestrogen deficiency, because it is primarily driven by SHOX gene haploinsufficiency (see genetic basis section).
• Cubitus valgus — increased carrying angle of the elbow; a minor dysmorphic feature.
• Short fourth metacarpal — knuckle sign positive.
• High-arched palate, multiple pigmented naevi.
Adolescent presentation:
• Primary amenorrhoea and absent pubertal development — the presenting complaint in approximately 30% of cases. Without oestrogen therapy, puberty does not occur because the streak gonads produce no oestrogen.
• Infertility — nearly universal in 45,X; a minority (~2–5%) of mosaic Turner have spontaneous puberty and rare spontaneous pregnancies.
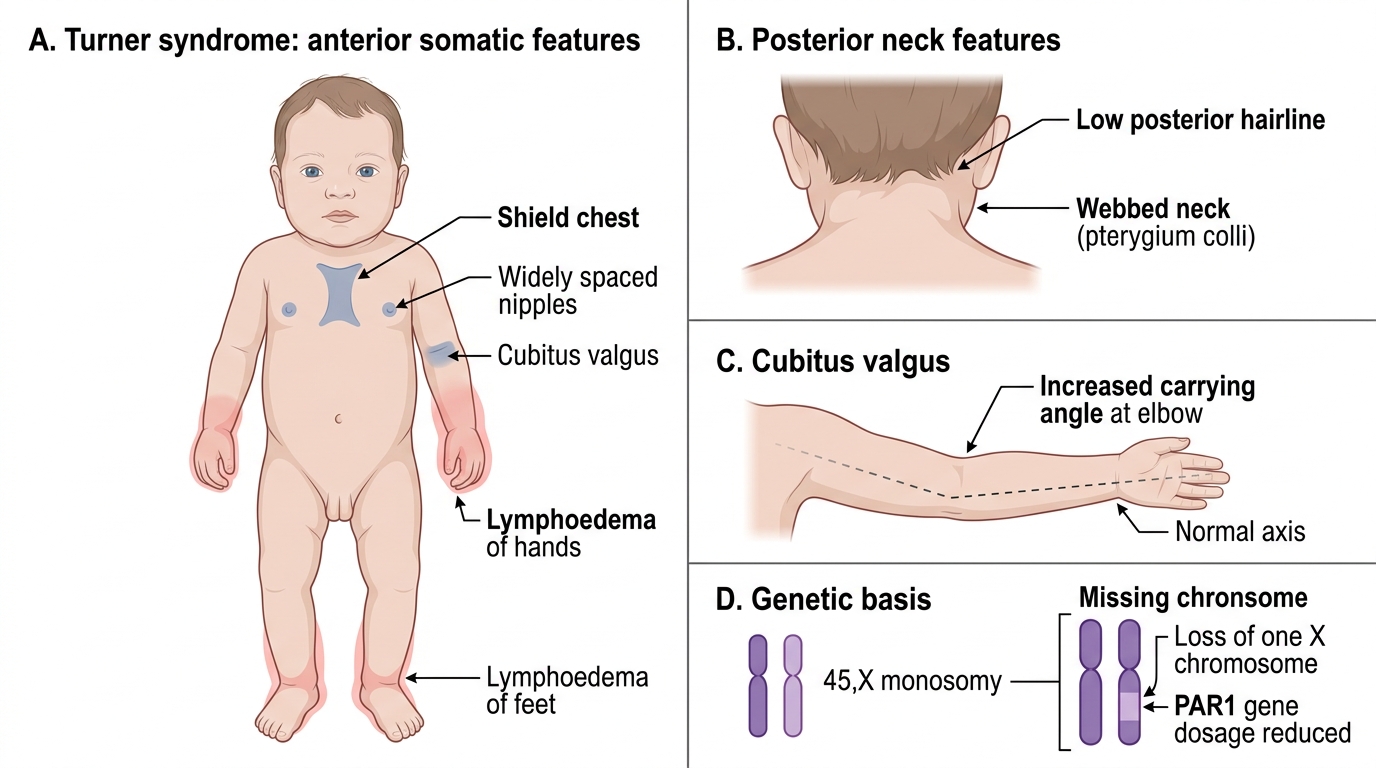
Somatic Features of Turner Syndrome
Genetic Basis and Pathophysiology
Turner syndrome results from complete or partial monosomy of the X chromosome. Unlike autosomal monosomies (which are invariably lethal), 45,X is viable because the majority of X-linked genes are already silenced on the second X chromosome in normal females via X-inactivation. However, genes in the pseudoautosomal region 1 (PAR1) of the X chromosome escape inactivation and are normally present in two copies in both males (Xp + Yp) and females (Xp + Xp). Loss of one PAR1 region — as in 45,X — disrupts gene dosage for these PAR1 genes, and this is the primary mechanism of short stature and skeletal abnormalities.
Cytogenetic variants:
1. Monosomy 45,X (~50% of Turner syndrome cases)
The karyotype is 45,X — only one sex chromosome is present. This is the classic Turner karyotype and typically produces the full somatic phenotype. The missing X is of paternal origin in approximately 75% of cases. The mechanism is non-disjunction or chromosome loss during early cell division.
2. Mosaicism: 45,X/46,XX or 45,X/46,XY (~30–40% of cases)
Post-zygotic chromosome loss creates two cell lines. In 45,X/46,XX mosaicism, the 46,XX cell line partially rescues the phenotype — features are milder, some patients have spontaneous puberty, and short stature may be less severe. In 45,X/46,XY mosaicism (rare), there is a risk of gonadoblastoma in the Y-bearing gonadal cells; prophylactic gonadectomy is recommended.
3. Structural X abnormalities (~10–20% of cases)
• Isochromosome Xq (46,X,i(Xq)) — the most common structural variant; loss of the short arm with duplication of the long arm; short stature prominent.
• Ring X chromosome and deletions of Xp are less common.
Pathophysiology of key features:
• Short stature: SHOX (Short stature homeobox) gene on PAR1 — haploinsufficiency causes disproportionate short stature, mesomelia, Madelung deformity (in Léri-Weill syndrome and Turner). This is why short stature occurs even in mosaic Turner and before oestrogen deficiency develops.
• Gonadal dysgenesis: The second X chromosome is needed for maintenance of primordial follicles; without it, follicles undergo accelerated atresia during fetal life, leaving only streak gonads (fibrous remnants) by late fetal development. Result: primary gonadal failure, absent endogenous oestrogen, primary amenorrhoea.
• Cardiac defects: Bicuspid aortic valve and aortic coarctation arise from abnormal lymphatic-vascular development during embryogenesis, likely related to the same X-linked gene-dosage effects responsible for the lymphoedema.
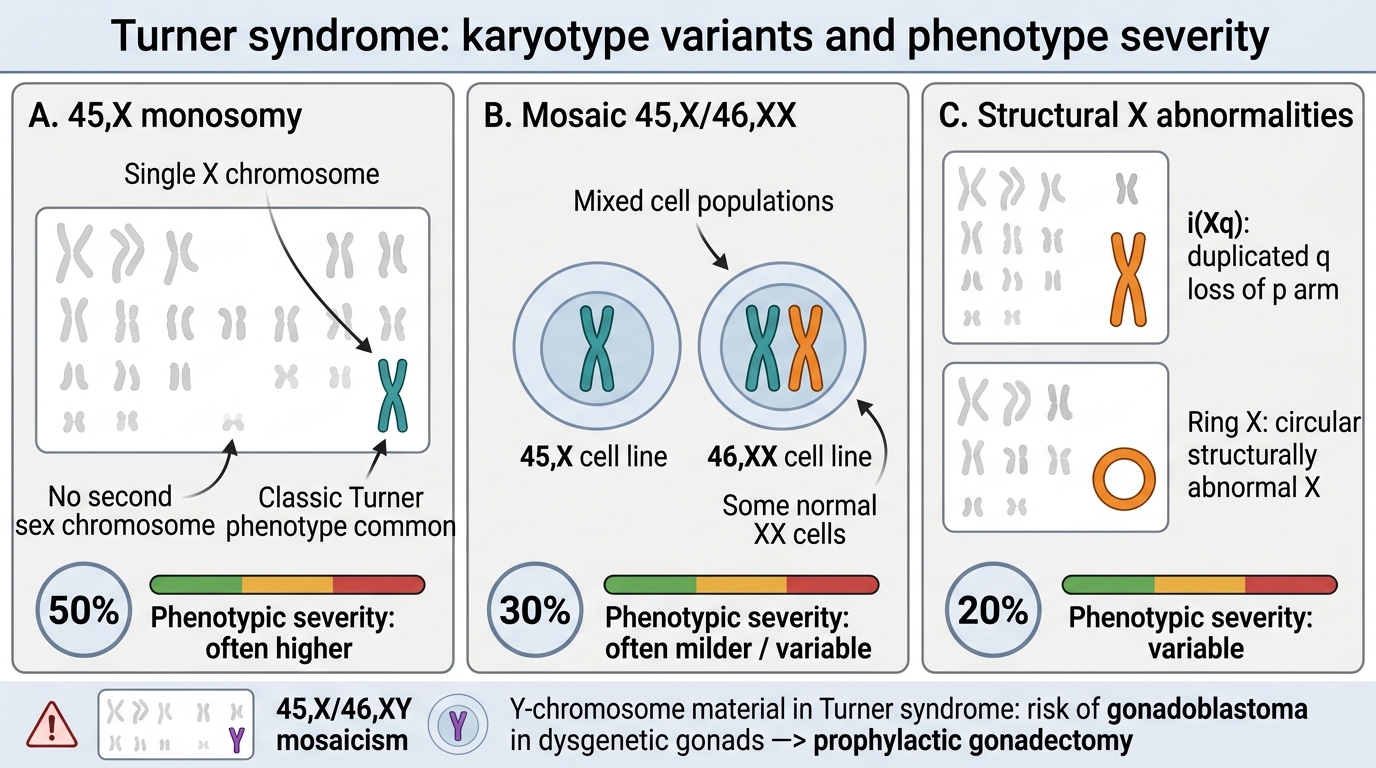
Turner Syndrome Karyotype Variants
SELF-CHECK
A girl with Turner syndrome has a karyotype of 45,X/46,XY mosaic. Apart from the usual Turner syndrome surveillance, what additional management is required specifically because of the XY cell line?
A. Additional doses of growth hormone because XY mosaics are taller
B. Prophylactic gonadectomy due to risk of gonadoblastoma in Y-bearing gonadal cells
C. Testosterone replacement instead of oestrogen replacement
D. No additional management — 45,X/46,XY mosaics have a normal life expectancy
Reveal Answer
Answer: B. Prophylactic gonadectomy due to risk of gonadoblastoma in Y-bearing gonadal cells
In 45,X/46,XY mosaicism, the Y-bearing cell line confers a significant risk of gonadoblastoma — a tumour arising in dysgenetic gonads that contain Y-chromosome material. Gonadoblastoma itself is usually benign but can undergo malignant transformation to dysgerminoma. Prophylactic gonadectomy (removal of the streak gonads) is therefore recommended in all 45,X/46,XY Turner syndrome patients. These patients still require oestrogen replacement (not testosterone, as they are phenotypic females with gonadal dysgenesis). They do not receive extra growth hormone on account of the XY karyotype.
Diagnosis and Investigation
The diagnosis of Turner syndrome is confirmed by karyotype, but the clinical context determines when and how to suspect it. A systematic investigation panel serves two purposes: confirming the diagnosis and identifying the associated conditions that require immediate or ongoing management. The investigation approach differs depending on whether the presentation is neonatal (lymphoedema and dysmorphic features), paediatric (short stature on growth surveillance), or adolescent (pubertal delay and primary amenorrhoea). In each context, karyotype is the essential first investigation once Turner syndrome is suspected; additional investigations then map the extent of organ involvement. It is important to investigate systematically even when the diagnosis seems clinically obvious — an apparently isolated somatic presentation may conceal a significant bicuspid aortic valve or a horseshoe kidney that will influence management. The hormonal profile provides further support for the diagnosis and guides the timing of hormone replacement therapy.
Karyotype:
• Peripheral blood karyotype is the confirmatory investigation. At least 30 cells should be examined to exclude mosaicism (low-level mosaics may be missed on smaller counts).
• Karyotype identifies the specific variant: 45,X, mosaic 45,X/46,XX, 45,X/46,XY, or structural X abnormality.
• Prenatal diagnosis: cfDNA screening detects sex-chromosome aneuploidies from 10 weeks with high sensitivity; confirmation by amniocentesis or CVS provides a definitive karyotype.
Hormonal profile:
• FSH and LH: Markedly elevated in 45,X Turner (hypergonadotropic hypogonadism) — the hallmark biochemical finding. In infancy and early puberty, FSH is elevated; it is the earliest hormonal signal of gonadal failure.
• Oestradiol: Very low or undetectable.
• Serum IGF-1 and IGFBP-3: To assess GH axis and monitor GH therapy.
Cardiac investigations:
• Echocardiography (all newly diagnosed patients): to identify bicuspid aortic valve, aortic coarctation, aortic dilation, and other structural defects.
• Blood pressure in all four limbs: Radio-femoral delay + upper limb hypertension + diminished femoral pulses = coarctation until proven otherwise.
• MRI of the aorta: At diagnosis and every 5–10 years thereafter for aortic dilation monitoring (Endocrine Society guidelines).
Other investigations:
• Renal ultrasound: To detect horseshoe kidney, duplex collecting system, and rotational anomalies (~30% of Turner syndrome patients have renal structural abnormalities).
• Thyroid function (TSH, anti-TPO antibodies): Autoimmune thyroiditis (Hashimoto's) occurs in ~30–50% of Turner syndrome over time; annual TSH from age 4.
• Bone age X-ray: To guide timing of GH and oestrogen therapy.
• Hearing assessment: Sensorineural hearing loss more common.
• Ophthalmology: Strabismus, ptosis assessment.
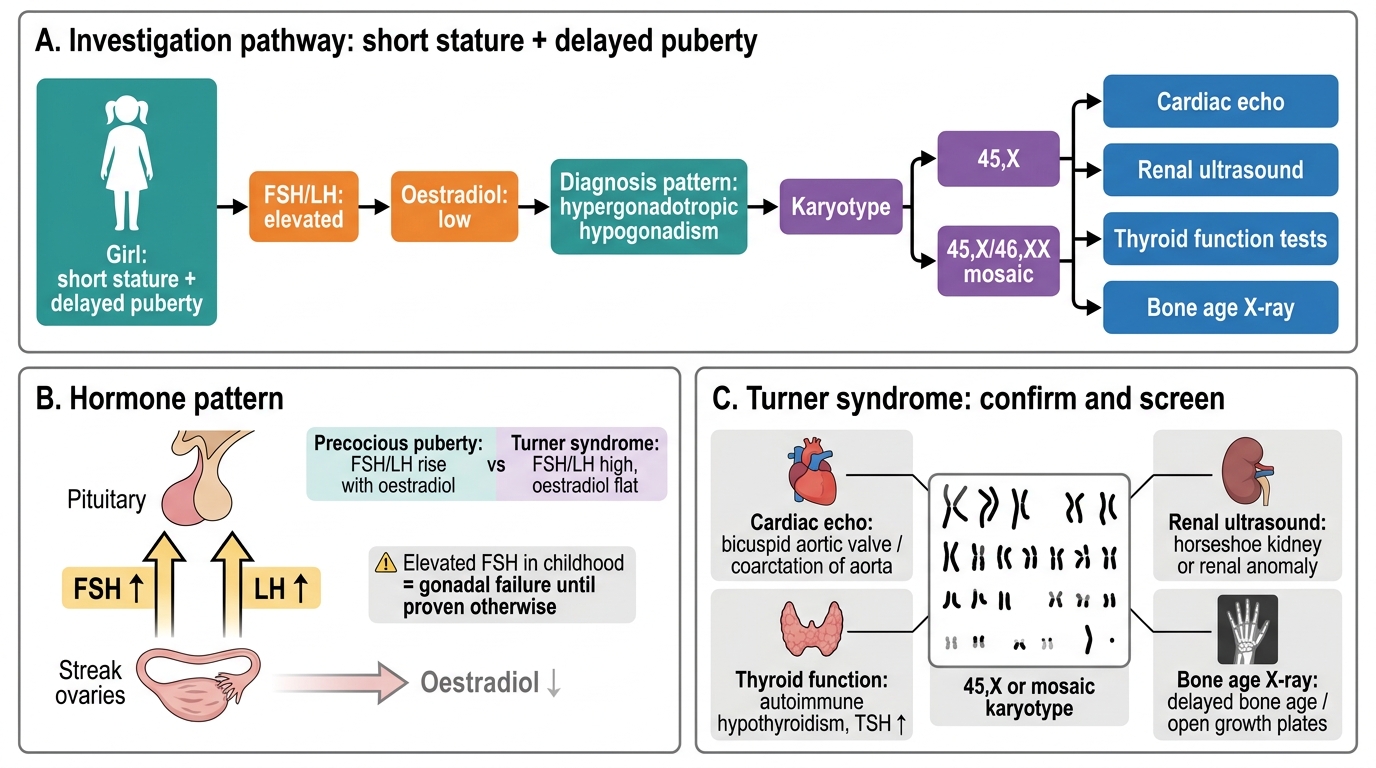
Turner Syndrome Investigation Pathway
CLINICAL PEARL
Elevated FSH in childhood = gonadal failure until proven otherwise. In normal prepubertal girls, FSH levels are low and LH is virtually undetectable. If a child presenting with short stature or pubertal delay is found to have markedly elevated FSH and LH with low oestradiol, this is hypergonadotropic hypogonadism — the pituitary is flooding the circulation with gonadotropins because the gonads are not responding. Turner syndrome is the most common cause in phenotypic females. Do not mistake an elevated FSH as a sign of precocious puberty — in true precocious puberty, FSH and LH rise in a coordinated, pulsatile pattern alongside oestradiol; in Turner, oestradiol is flat.