Page 5 of 14
PE29.4 | Turner Syndrome — SDL Guide (Part 2)
Management, Complications, and Genetic Counselling
Management of Turner syndrome is lifelong, multidisciplinary, and time-sensitive in several domains — growth hormone must be started before the growth window closes, oestrogen must be initiated at the right bone age to induce puberty and protect bone, and cardiac surveillance must be regular to detect aortic dilation before dissection. The endocrinologist, cardiologist, and gynaecologist share the long-term care, while the paediatrician coordinates and explains the plan to the family. The therapeutic goals are clear: maximise final adult height with GH therapy; replace the oestrogen that the streak gonads cannot produce; protect the heart and aorta from the cardiovascular complications that are the leading cause of premature mortality in Turner syndrome; and preserve bone density and quality of life across the lifespan. Genetic counselling is a distinct and important component, addressing both fertility options and the negligible recurrence risk for subsequent pregnancies.
Growth hormone therapy:
• Recombinant human GH (rhGH) is the standard of care for short stature in Turner syndrome, regardless of whether GH deficiency is documented on testing.
• Dose: ~0.05–0.067 mg/kg/day subcutaneous injection (higher than GH deficiency dose because SHOX haploinsufficiency does not respond to GH-deficiency dosing alone).
• Timing: Ideally started as soon as height falls below the normal centile, typically age 2–4 years; continuing until final height is achieved.
• Target: Most patients gain an extra 5–8 cm in final adult height with GH therapy compared to untreated Turner syndrome.
Oestrogen replacement (pubertal induction):
• In girls who do not have spontaneous puberty (the majority), oestrogen replacement is initiated to induce secondary sex characteristics, promote uterine growth, and protect bone mineral density.
• Timing: Initiated at ~11–12 years (bone age ~11 years); low-dose transdermal or oral oestradiol, escalated gradually over 2–3 years to mimic natural pubertal progression.
• Cyclic progesterone is added once breakthrough bleeding occurs or after 2 years of oestrogen, to protect the endometrium.
• HRT is continued until the natural age of menopause (~50 years).
Cardiac complications and surveillance:
• Bicuspid aortic valve (~30% of patients): Risk of aortic stenosis and regurgitation in adulthood; requires echocardiographic follow-up.
• Aortic coarctation (~10–15%): Presents with radio-femoral delay and upper limb hypertension; corrected surgically or by balloon dilation + stenting. Residual hypertension common — requires antihypertensive therapy.
• Aortic dilation and dissection: The most serious cardiovascular risk; MRI every 5–10 years. Risk factors: bicuspid valve, hypertension, coarctation, prior cardiac surgery, pregnancy.
Other complications:
• Hypothyroidism: Annual TSH from age 4; Hashimoto's thyroiditis is common (~30–50% of adults with Turner).
• Renal: Horseshoe kidney — usually asymptomatic but increases UTI and stone risk; monitor accordingly.
• Osteoporosis: Oestrogen deficiency and GH deficiency both reduce bone density; adequate HRT and calcium/vitamin D supplementation are protective.
• Neurocognitive: Normal intelligence overall, but specific difficulties with visual-spatial tasks and social cognition; educational support may be needed.
Genetic counselling:
• Recurrence risk: Turner syndrome is sporadic; recurrence risk for future pregnancies is not significantly elevated above background. Parental karyotypes are normal.
• Fertility: Most women with 45,X Turner are infertile due to streak gonads. Oocyte donation + IVF is the main option for biological parenthood. Women with mosaic Turner who have spontaneous cycles may attempt natural conception but carry a higher risk of 45,X offspring and spontaneous abortion.
• Antenatal genetic testing (cfDNA, amniocentesis) should be offered in any future pregnancy.
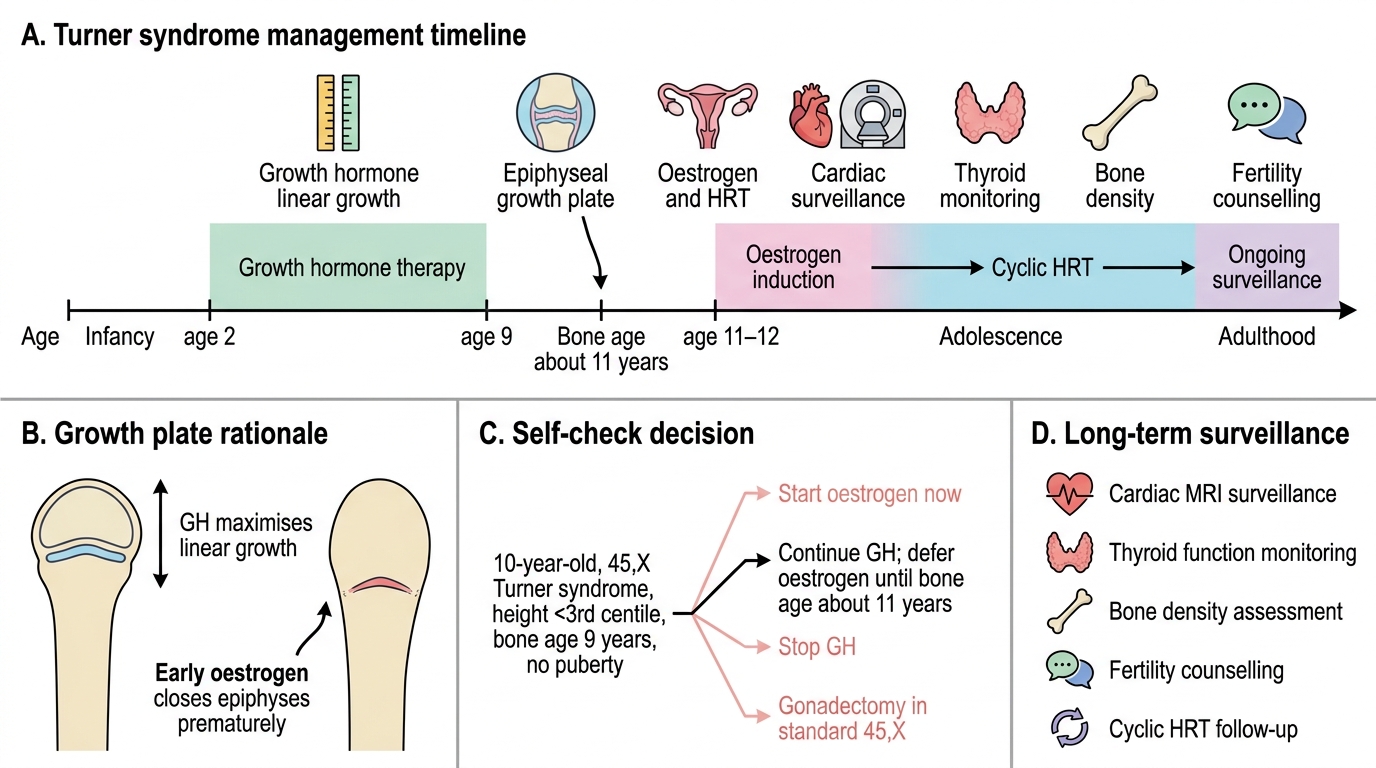
Turner Syndrome Management Timeline
SELF-CHECK
A 10-year-old girl with confirmed Turner syndrome (45,X) has a bone age of 9 years and a height of 128 cm (well below 3rd centile). She has no signs of puberty. Which combination of interventions is most appropriate at this time?
A. Start oestrogen replacement now at age 10 to induce puberty as soon as possible
B. Continue growth hormone therapy; defer oestrogen until bone age reaches ~11 years
C. Stop growth hormone since she has not yet responded after several years
D. Refer for gonadectomy; then start both GH and oestrogen simultaneously
Reveal Answer
Answer: B. Continue growth hormone therapy; defer oestrogen until bone age reaches ~11 years
In Turner syndrome management, growth hormone and oestrogen replacement are intentionally staggered. GH therapy should be continued (or started if not already) to maximise linear growth. Oestrogen replacement is deferred until bone age reaches approximately 11 years — initiating oestrogen too early closes the epiphyses prematurely and sacrifices final adult height. The bone age of 9 years means there is still significant growth potential; oestrogen induction should wait until bone age ~11. Gonadectomy is only indicated in 45,X/46,XY mosaicism, not in standard 45,X Turner.
Self-Assessment
Use these questions to assess your readiness to manage a girl with Turner syndrome in the clinical setting. Turner syndrome is a multi-system condition where missing or delaying any component of the management plan has a measurable impact on adult outcomes — height, cardiac health, bone density, and quality of life. The questions below test whether you can integrate the genetic, endocrinological, cardiac, and counselling dimensions of this condition into a coherent clinical plan. A student who can answer all six questions below without reference to notes has demonstrated the competency level expected of PE29.4. Work through each question carefully as if it were a viva or OSCE station, and note any knowledge gaps for targeted review.
Structured self-check questions:
- A neonate has lymphoedema of both hands and a low posterior hairline. What is your initial investigation, and if the karyotype returns 45,X, what three specialist referrals will you make within the first month of life?
- Explain why a girl with Turner syndrome has short stature before gonadal failure has had time to deplete her oestrogen. Name the specific gene involved.
- What is the expected gonadotropin profile in a 15-year-old with Turner syndrome presenting with primary amenorrhoea? Distinguish this from central (hypothalamic/pituitary) causes.
- At what bone age would you initiate oestrogen replacement, and why is timing critical for final height?
- A woman with Turner syndrome (45,X) wishes to conceive. What options exist, and what antenatal testing would you recommend?
- List four conditions requiring regular surveillance in Turner syndrome, with the recommended investigation and frequency for each.
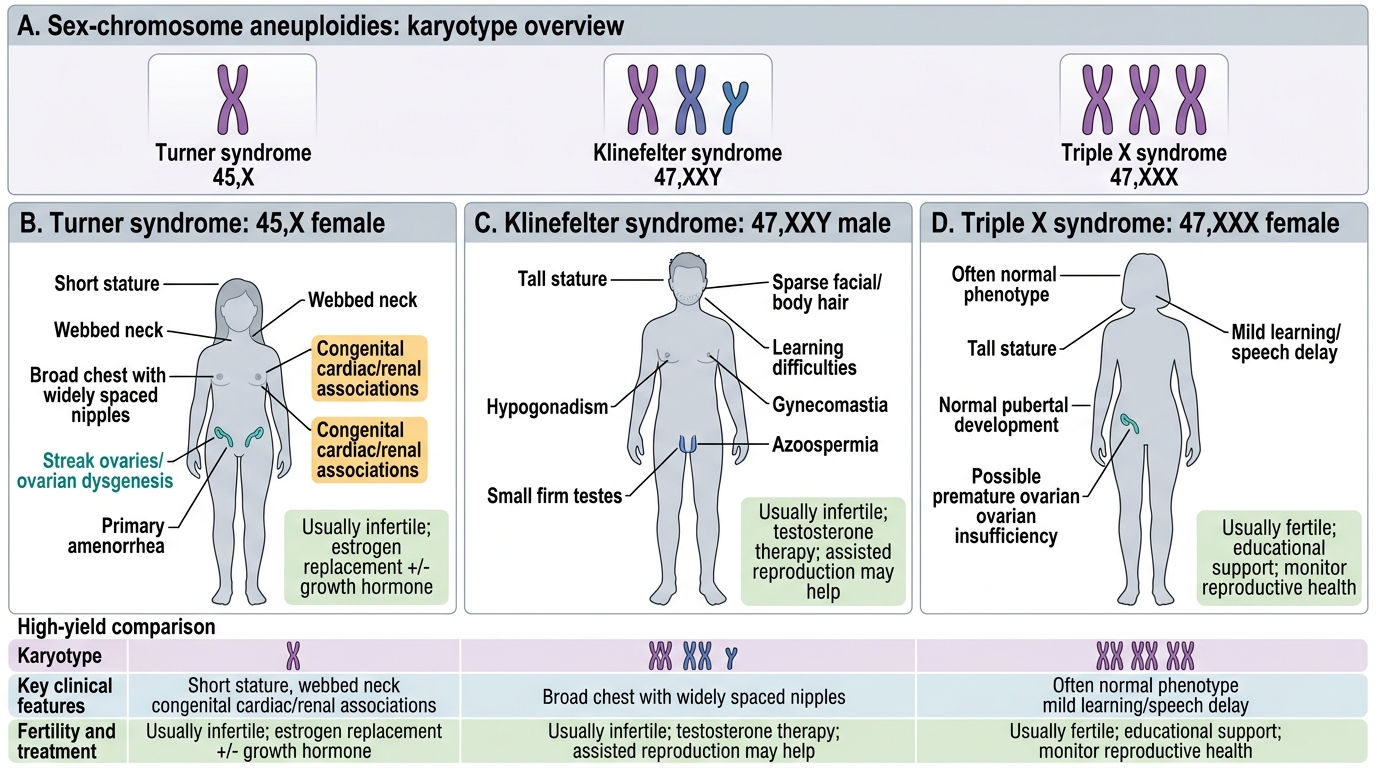
Sex-Chromosome Aneuploidies: Turner, Klinefelter, and Triple X