Page 6 of 20
PA32.2-3 | Bone & Soft Tissue Tumors — SDL Guide
Learning Objectives
- Classify bone tumors by matrix type and cell of origin (bone-forming, cartilage-forming, fibrous, vascular, marrow-derived) and distinguish benign from malignant variants
- Describe the etiology, pathogenesis, radiologic features (X-ray, MRI), gross and microscopic morphology, and complications of osteosarcoma, osteochondroma, chondrosarcoma, giant cell tumor, Ewing sarcoma, and metastatic bone disease
- Classify soft tissue tumors (lipoma/liposarcoma, rhabdomyosarcoma, leiomyosarcoma, fibrosarcoma, synovial sarcoma) and apply general principles of grading, staging, and routes of spread
- Identify the most common benign and malignant bone tumors, recognize site predilections by age, and correlate clinical and radiologic findings with histopathology for each major tumor
INSTRUCTIONS
Bone and soft tissue tumors are among the highest-yield topics in Year-2 Pathology — expect spot diagnoses on X-rays and photomicrographs in university examinations, and recognize them in surgical oncology and orthopedics postings. Every tumor described here carries a distinct age group, skeletal location, radiologic sign, and histologic hallmark. Learn these four columns as a table in your mind. Work through the blocks sequentially; each micro-quiz appears immediately after the content it tests.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch. 26 (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 15-year-old boy walks into the orthopedic OPD with a two-month history of pain around his left knee that wakes him at night. His mother assumed it was 'growing pains.' The X-ray taken on a whim shows something that stops the resident cold — a dense, cloudlike mass in the distal femur, with a triangle of raised periosteum at the edge and a burst of bony spicules shooting outward from the cortex like rays of the sun. The radiologist circles it and writes three words: Rule out osteosarcoma. Within 72 hours, this boy is at a tertiary cancer centre. Why did that X-ray raise an alarm? What is that periosteal triangle? Why does this tumor appear in teenagers around the knee? By the end of this module, you will read that X-ray the way the resident did — and understand exactly why those two words changed everything.
WHY THIS MATTERS
Bone and soft tissue tumors span two competencies — PA32.2 and PA32.3 — and they appear in every format of your university examination: spot X-rays, photomicrographs, long SAQs, and OSPE stations. In clinical postings you will encounter these patients in orthopedics, surgical oncology, and pediatrics. You will be expected to recognize the age, site, and radiologic pattern of each tumor instantly, correlate them with the biopsy findings, and counsel patients about prognosis. Beyond exams, osteosarcoma is the most common primary bone malignancy in adolescents and young adults — a demographic you will serve throughout your career. Understanding metastatic bone disease is equally critical: it is the most common malignant bone disease overall, and nearly every patient admitted with advanced breast, prostate, or lung cancer will have bony involvement. This module builds the vocabulary you need for both the examination hall and the ward.
RECALL
Before you start, activate what you already know. From Anatomy and Physiology you know that bone is a living connective tissue with osteoblasts (bone-forming cells) and osteoclasts (bone-resorbing cells). You know that long bones have a diaphysis (shaft), metaphysis (growth-plate region), and epiphysis (articular end). From General Pathology (PA31) you know that neoplasia is uncontrolled growth, that tumors are classified as benign or malignant, and that malignant tumors metastasize. You have also learned that tumor suppressor genes like RB1 and TP53 act as brakes on the cell cycle — when these brakes fail, malignancy follows. Bring all of this forward: in this module you will see exactly how osteoblasts, osteoclasts, and chondrocytes give rise to distinct tumor families, and how the same RB and p53 pathways whose loss you studied in general pathology drive the most dangerous bone cancer in young people.
Overview: Classifying Bone Tumors
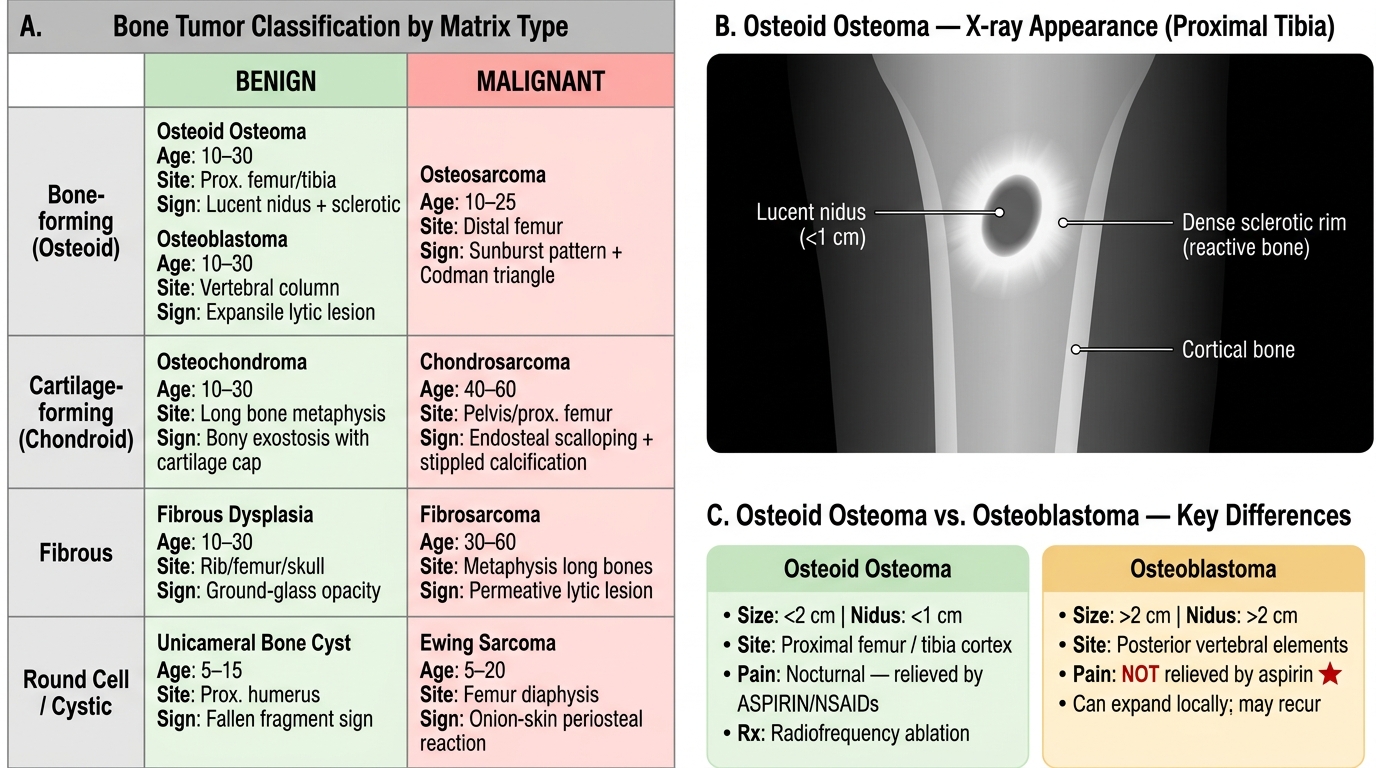
Bone tumors are classified by the matrix they produce (or the cell they resemble), which determines both their name and their behavior. This is the master table you need to carry in your head:
| Tumor type | Benign | Malignant | Peak age | Usual site |
|---|---|---|---|---|
| Bone-forming | Osteoid osteoma, Osteoblastoma | Osteosarcoma | Teens–young adults | Metaphysis of long bones |
| Cartilage-forming | Osteochondroma, Enchondroma | Chondrosarcoma | Varies (see below) | Metaphysis / medullary cavity |
| Unknown/osteoclast | Giant cell tumor | (Locally aggressive) | 20–40 yrs | Epiphysis |
| Marrow/neural | — | Ewing sarcoma | Children / teens | Diaphysis |
| Metastatic | — | (Secondary) | Adults >40 | Axial skeleton, proximal femur |
| Fibrous | Fibrous dysplasia | Fibrosarcoma | Adults | Metaphysis / diaphysis |
| Soft tissue — fat | Lipoma | Liposarcoma | Adults | Extremities, retroperitoneum |
| Soft tissue — muscle | — | Rhabdomyosarcoma, Leiomyosarcoma | Children / adults | Extremities, retroperitoneum |
Three general rules hold for almost every bone tumor:
1. Benign tumors are slow-growing, have sharp radiologic margins (sclerotic rim), rarely cause systemic symptoms.
2. Malignant tumors destroy cortex, invoke periosteal reactions, extend into soft tissue, and metastasize — primarily hematogenously to the lungs.
3. Periosteal reactions on X-ray are key: a smooth, layered reaction suggests slow growth; a Codman triangle or sunburst pattern signals rapid, aggressive growth.
Bone Tumor Classification by Matrix Type with Osteoid Osteoma Radiologic Sign
Osteoid Osteoma and Osteoblastoma
Osteoid osteoma and osteoblastoma are benign bone-forming tumors that differ mainly in size and symptoms.
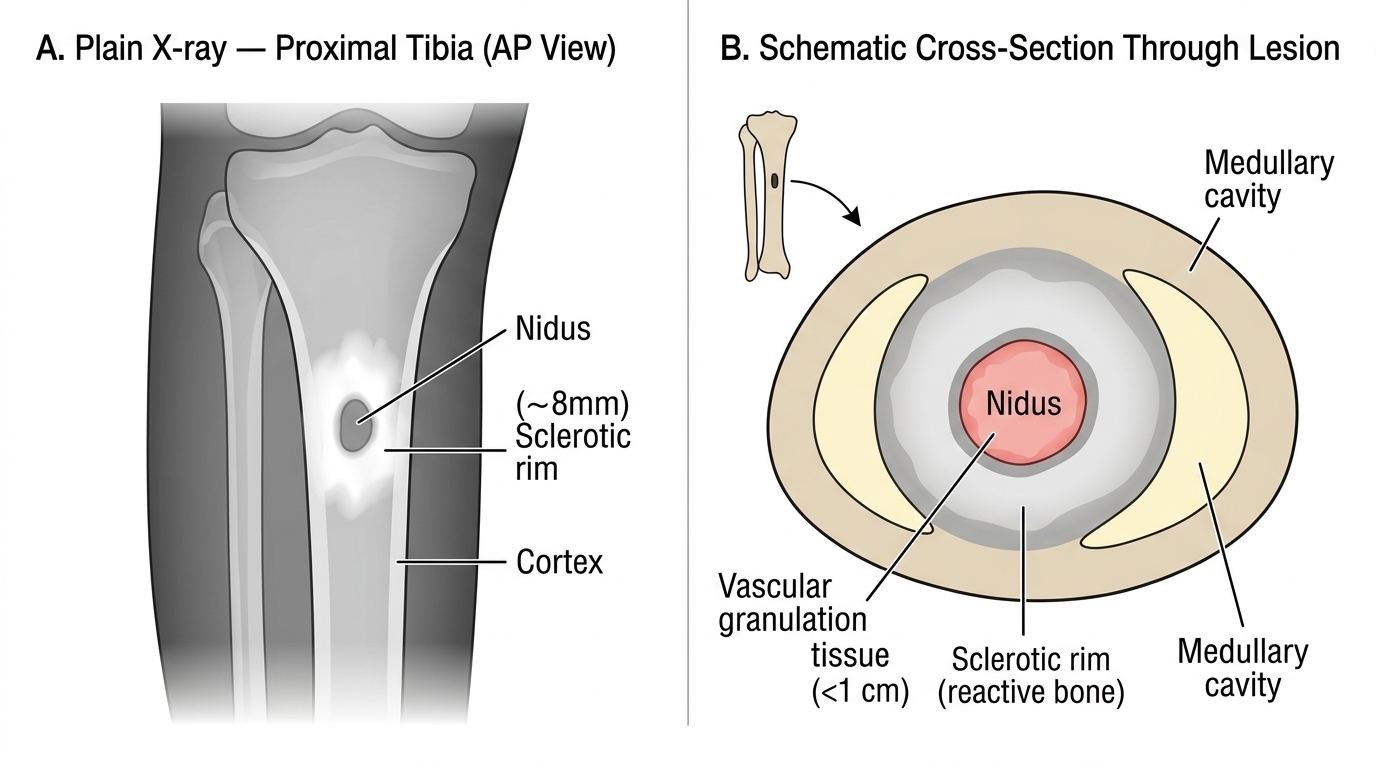
Osteoid osteoma
- Small (<2 cm), forms a central radiolucent nidus of woven osteoid rimmed by dense reactive sclerosis.
- Classic presentation: nocturnal pain that is dramatically relieved by aspirin or NSAIDs — due to prostaglandin production by the nidus.
- Sites: cortex of the proximal femur and tibia (diaphysis or metaphysis).
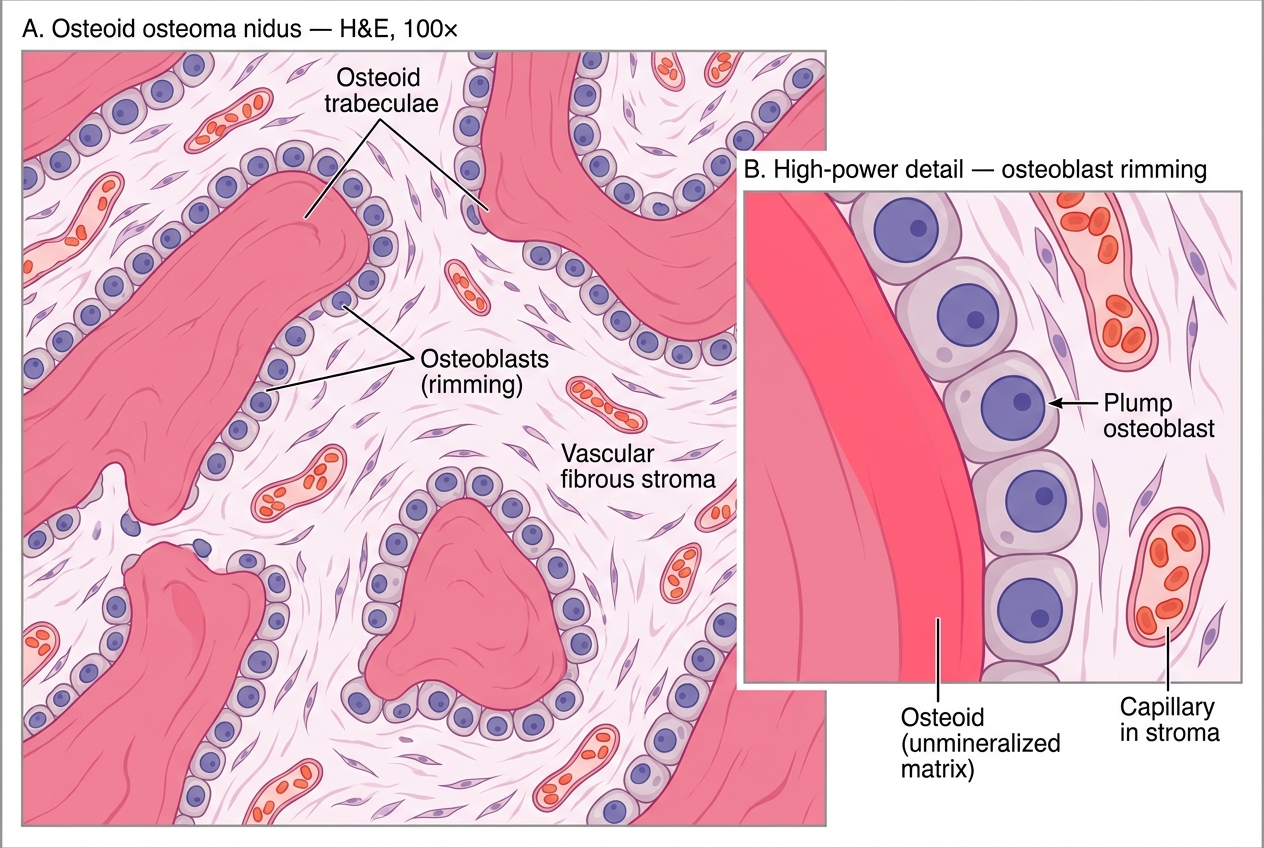
- Histology: tangled woven osteoid trabeculae lined by a single layer of osteoblasts, set in a vascular fibrous stroma. No significant atypia.
- Radiologic sign: lucent nidus (<1 cm) surrounded by dense bony sclerosis — the halo sign.
- Treatment: radiofrequency ablation (CT-guided); rarely resection.
Osteoblastoma ("giant osteoid osteoma")
- Larger (>2 cm), often in the posterior elements of the vertebral column (unlike osteoid osteoma).
- Pain is NOT relieved by aspirin — a key clinical distinguishing point.
- Histology identical to osteoid osteoma but larger nidus; can locally expand and even recur.
- No malignant potential in the classic form.
Osteoid Osteoma — Radiographic Appearance and Lesion Architecture
Histology of Osteoid Osteoma Nidus (H&E)
Osteosarcoma — The High-Yield Malignant Bone Tumor
Osteosarcoma (osteogenic sarcoma) is the most common primary malignant tumor of bone (excluding multiple myeloma and metastases). It demands detailed knowledge for your examination.
Epidemiology and etiology
- Peak incidence: 10–20 years (second decade), with a smaller second peak in elderly (often Paget-related).
- Male > female (1.5:1).
- Sites: metaphysis of long bones around the knee — distal femur (~40%), proximal tibia (~20%), proximal humerus (~10%). The distal femur metaphysis is the single most common site.
- Etiology: sporadic in most cases. Genetic factors — loss of RB1 (retinoblastoma gene, 13q14) and TP53 mutation are the major drivers. Children with hereditary retinoblastoma have a markedly elevated risk (RB1 loss in both alleles).
- Paget's disease of bone, prior radiation are risk factors for secondary osteosarcoma in older adults.
Pathogenesis
Osteoblast precursors accumulate RB1 and TP53 mutations (spontaneous or radiation-induced) → loss of cell cycle checkpoints → uncontrolled osteoblast proliferation → production of malignant osteoid directly by tumor cells (this is the defining feature: osteoid produced by sarcomatous stroma).
Clinical features
- Deep, persistent pain increasing in severity; not relieved by rest or NSAIDs.
- Swelling, tenderness, and warmth over the metaphysis.
- Pathological fracture (late, uncommonly).
- No fever (distinguishes from Ewing sarcoma/osteomyelitis).
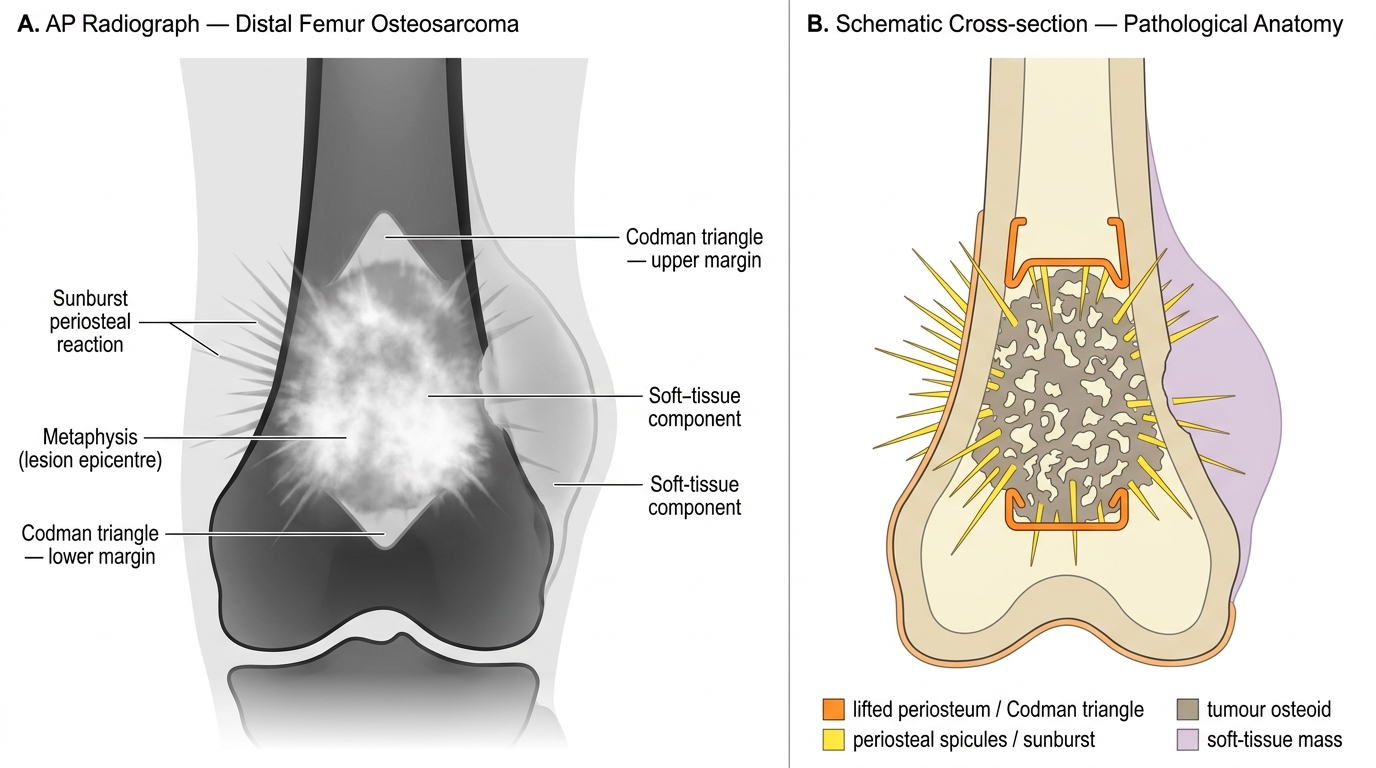
Radiologic features (MUST KNOW)
- Mixed lytic and sclerotic lesion in the metaphysis — the tumor is making bone and destroying it simultaneously.
- Codman triangle: as the tumor elevates the periosteum, a triangular shell of reactive new bone forms at the lifting edge of the periosteum. Seen at the proximal and distal margins of the tumor on X-ray.
- Sunburst (sunray) pattern: tumor bone grows radially outward through the elevated periosteum, creating spicules that radiate from the cortex like rays of the sun.
- Cortical destruction and soft tissue extension are common.
- MRI: best for defining extent of medullary involvement and skip lesions (small satellite tumor foci separated from the main mass).
Radiological and Pathological Anatomy of Distal Femur Osteosarcoma
Gross pathology
- Gray-white gritty mass replacing the metaphysis; may break through cortex into soft tissue.
- Hemorrhage and necrosis common in large tumors.
Histology (DEFINITIVE CRITERION)
- Malignant spindle cells (sarcomatous stroma) directly producing lace-like malignant osteoid (unmineralized bone matrix).
- Osteoid appears as eosinophilic, amorphous, homogeneous material deposited between atypical cells.
- Cells show marked nuclear pleomorphism, hyperchromasia, and mitotic figures (including atypical mitoses).
- Variants: osteoblastic (most common), chondroblastic (cartilage matrix), fibroblastic (fibrous stroma).
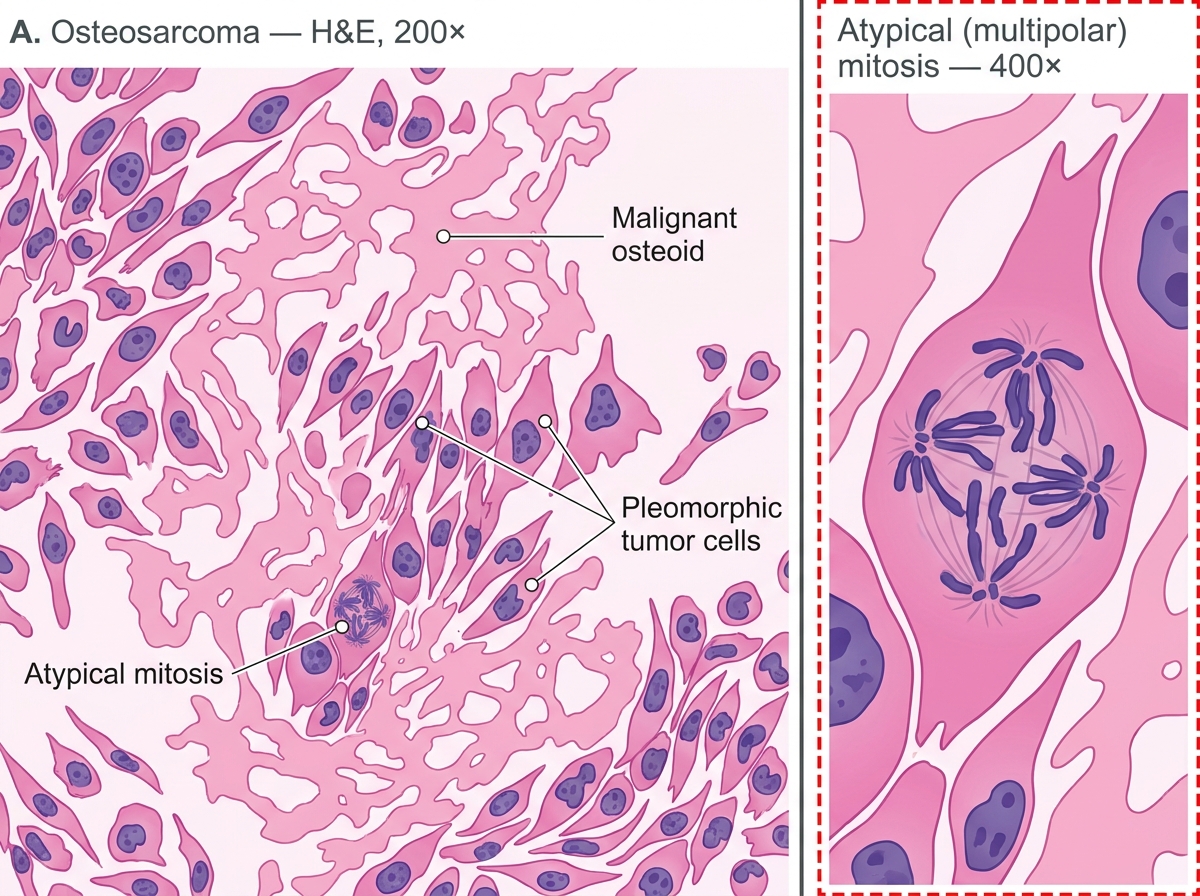
Osteosarcoma — H&E Photomicrograph (200× with 400× inset): Malignant Osteoid and Pleomorphic Tumor Cells
Spread and prognosis
- Hematogenous spread to lungs in 80% if untreated — lung metastases appear as "cannonball" nodules on chest X-ray/CT.
- Lymph node spread is rare.
- Prognosis: With limb-salvage surgery + neoadjuvant/adjuvant chemotherapy (methotrexate, doxorubicin, cisplatin), 5-year survival reaches 65–70% for localized disease. Metastatic disease at presentation: <20% 5-year survival.
CLINICAL PEARL
Codman triangle ≠ osteosarcoma alone. The Codman triangle is a periosteal reaction to any rapidly growing lesion that lifts the periosteum. You can see it in Ewing sarcoma, aggressive osteomyelitis, and even metastatic disease. What makes osteosarcoma the most likely diagnosis is the combination of: (1) a teenager, (2) distal femur metaphysis, (3) Codman triangle plus sunburst, and (4) mixed lytic-sclerotic lesion. When all four align, the diagnosis is osteosarcoma until proved otherwise.
SELF-CHECK
A 16-year-old presents with 3 months of worsening knee pain. X-ray shows a mixed lytic-sclerotic metaphyseal lesion of the distal femur with a sunburst periosteal reaction. Histology reveals malignant spindle cells depositing lace-like eosinophilic matrix. Which molecular pathway is most commonly disrupted in this tumor's pathogenesis?
A. BCR-ABL translocation
B. Loss of RB1 and TP53 tumor suppressor function
C. EWSR1-FLI1 chromosomal translocation
D. KRAS point mutation
Reveal Answer
Answer: B. Loss of RB1 and TP53 tumor suppressor function
Osteosarcoma is driven primarily by loss of the RB1 (retinoblastoma) and TP53 tumor suppressor genes, which remove critical cell-cycle checkpoints in osteoblast precursors. BCR-ABL is the hallmark of CML. EWSR1-FLI1 t(11;22) is the defining translocation of Ewing sarcoma. KRAS mutations drive carcinomas of the pancreas, colon, and lung — not osteosarcoma.