Page 7 of 20
PA32.2-3 | Bone & Soft Tissue Tumors — SDL Guide (Part 2)
Cartilage-Forming Tumors: Osteochondroma, Enchondroma, Chondrosarcoma
Osteochondroma (exostosis)
- Most common benign bone tumor overall.
- A bony projection capped by cartilage that arises from the metaphysis, pointing away from the joint.
- Pathogenesis: displacement of a fragment of growth plate cartilage to the metaphyseal surface → continues to grow until skeletal maturity.
- Sites: distal femur, proximal tibia, proximal humerus metaphyses.
- Clinical: painless, hard lump near a joint; discovered incidentally or because of soft-tissue impingement.
- Radiologic sign: broad-based or pedunculated bony excrescence arising from the metaphyseal cortex, with medullary cavity continuous with the host bone's medullary canal.
- Histology: outer cartilage cap (hyaline cartilage), with enchondral ossification occurring at its base. Cap thickness >2 cm in an adult = suspect malignant transformation.
- Multiple hereditary exostoses (MHE): autosomal dominant, EXT1/EXT2 mutations; hundreds of osteochondromas; risk of chondrosarcoma transformation ~1–5%.
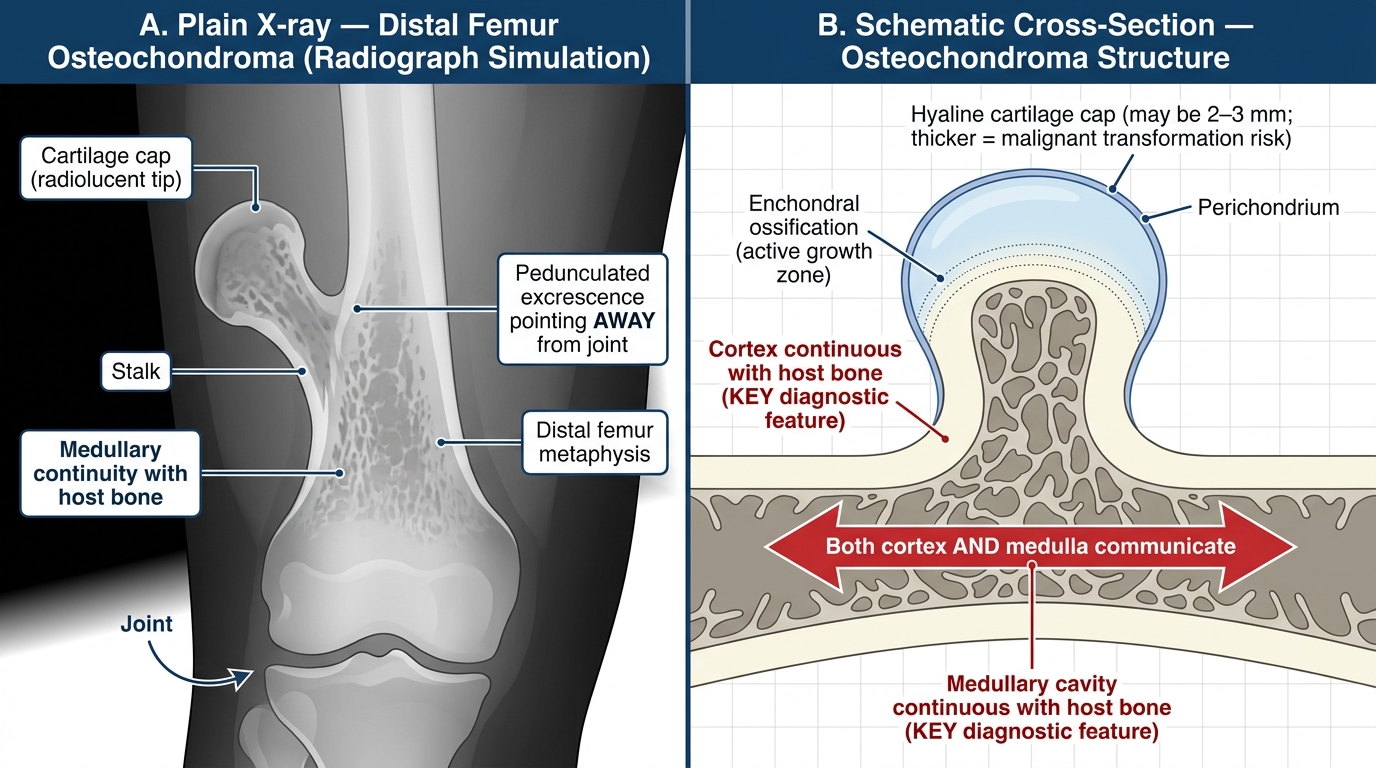
Osteochondroma of the Distal Femur — Radiological Appearance and Structural Anatomy
Enchondroma
- Benign intramedullary tumor of hyaline cartilage, typically in the medullary cavity of short tubular bones (phalanges of hands and feet).
- Usually solitary; Ollier disease = multiple enchondromas (no hereditary pattern); Maffucci syndrome = enchondromas + soft tissue hemangiomas (IDH1/IDH2 mutations; high malignant risk).
- Radiologic: oval lucency within the medullary cavity with "rings and arcs" or "popcorn" calcification.
- Histology: lobules of hypocellular hyaline cartilage with small uniform chondrocytes; no cytologic atypia.
- Most are incidental findings; symptomatic or enlarging lesions in adults warrant biopsy to exclude low-grade chondrosarcoma.
Chondrosarcoma
- Malignant tumor producing cartilaginous matrix — the second most common primary malignant bone tumor after osteosarcoma.
- Peak age: 40–70 years (older than osteosarcoma — this age contrast is exam-tested).
- Sites: central skeleton — pelvis, proximal femur, shoulder girdle, ribs. NOT typically in the distal extremities (unlike enchondroma).
- Most are low-grade (grade 1 or 2) and grow slowly; grade 3 chondrosarcomas are rare and aggressive.
- Etiology: can arise de novo (central chondrosarcoma) or in a pre-existing osteochondroma (peripheral chondrosarcoma).
- Radiologic: medullary lytic lesion with cortical thickening/endosteal scalloping, "rings and arcs" calcification (cartilage matrix calcification in annular pattern).
- Histology: lobules of malignant hyaline cartilage; chondrocytes show cytologic atypia — hypercellularity, binucleated cells, nuclear pleomorphism (compared to the bland uniform cells of enchondroma).
- Key molecular marker: IDH1/IDH2 mutations (same as gliomas) in ~50% of central chondrosarcomas.
- Treatment: surgical resection only — chondrosarcoma is resistant to radiotherapy and chemotherapy, which is a critical fact distinguishing it from osteosarcoma.
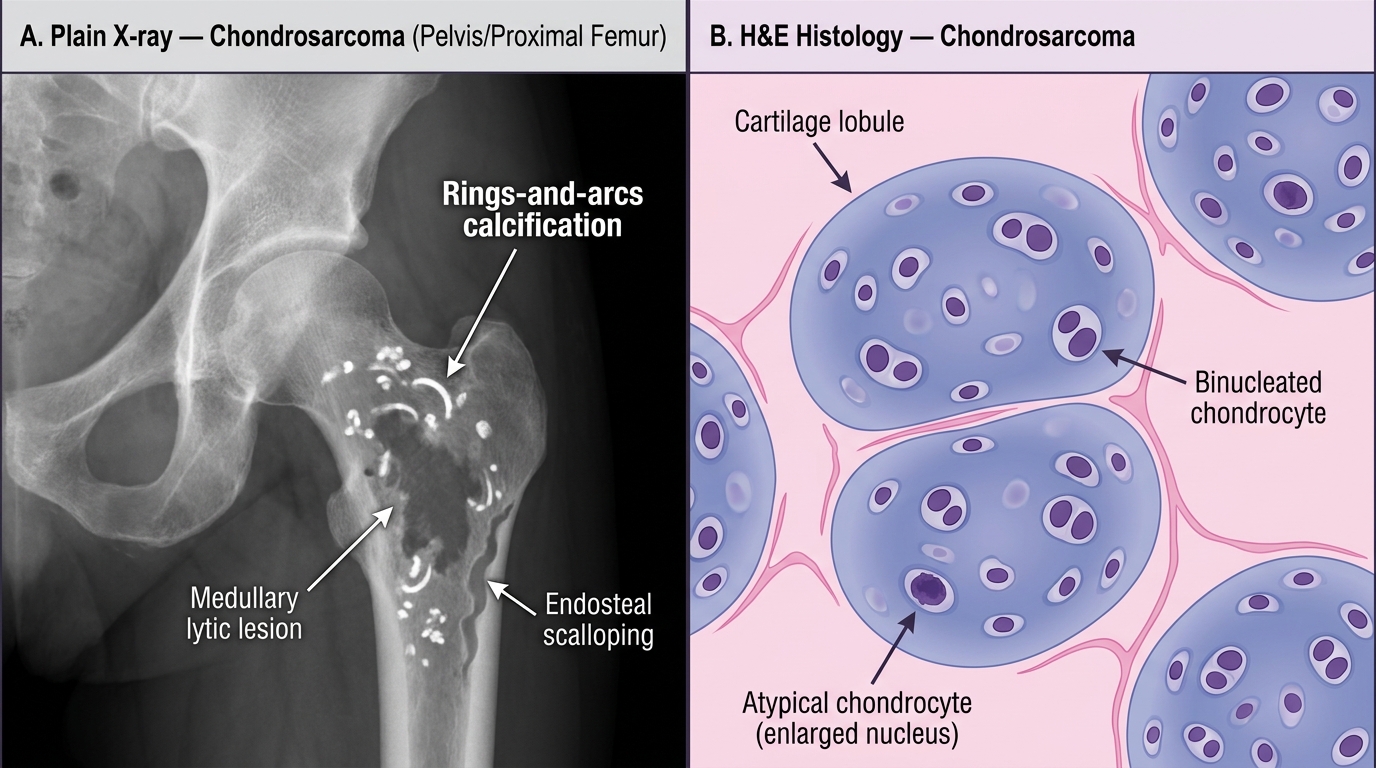
Chondrosarcoma: Radiological vs Histological Features
Giant Cell Tumor of Bone (Osteoclastoma)
Giant cell tumor of bone (GCT) is a locally aggressive, potentially malignant tumor with a distinctive biology and location that makes it a perennial examination favorite.
Key facts
- Age: 20–40 years (skeletally mature young adults — a critical distinction from osteosarcoma in teenagers).
- Site: the epiphysis — almost always arises in the epiphysis of long bones and extends up to and abuts the articular cartilage. Distal femur, proximal tibia, distal radius are most common. Osteosarcoma is in the metaphysis; GCT is in the epiphysis — this contrast is tested every year.
- Behavior: locally aggressive (can recur after curettage) and approximately 1–2% metastasize to the lungs (despite appearing histologically benign).
Radiology — "soap bubble" appearance
- Purely lytic lesion, no matrix mineralization.
- Eccentric, extends to the subchondral plate.
- "Soap bubble" (trabeculated lucency) pattern on X-ray — multiple rounded lucencies separated by thin bony septa, like soap bubbles.
- No sclerotic rim (distinguishes it from enchondroma); no periosteal reaction.
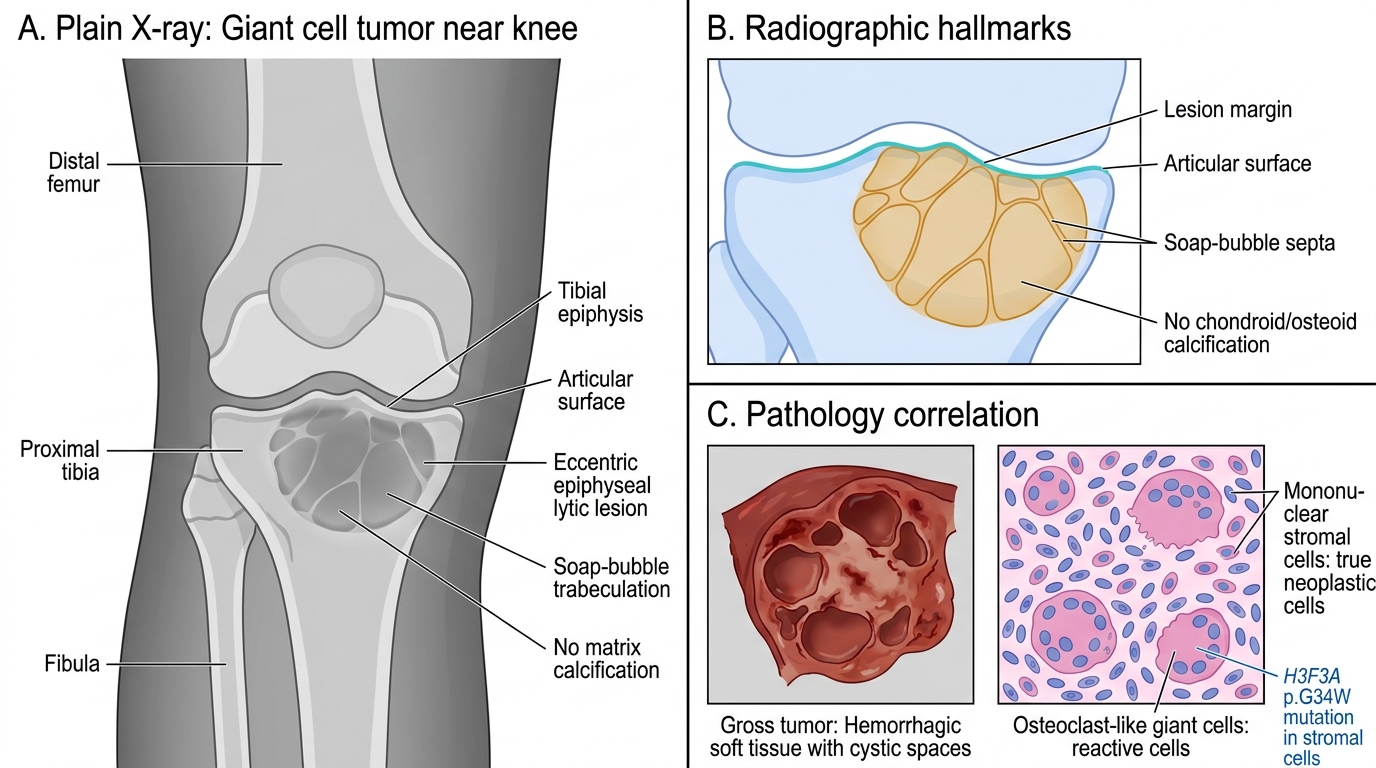
Giant Cell Tumor of Bone: X-ray Hallmarks
Pathology
- Gross: red-brown, hemorrhagic, soft tumor; may have cystic spaces.
- Histology (DEFINITIVE): two populations of cells:
1. Mononuclear stromal cells — oval/spindle-shaped with bland nuclei; these are the true neoplastic cells (they are the proliferating component).
2. Multinucleated giant cells — osteoclast-like (contain 50–100 nuclei), uniformly distributed throughout the tumor; nuclei are identical to those of the mononuclear stromal cells. These giant cells are reactive, not neoplastic.
- Molecular: H3F3A gene mutation (p.G34W) in the mononuclear stromal cells — detectable by immunohistochemistry and useful diagnostically.
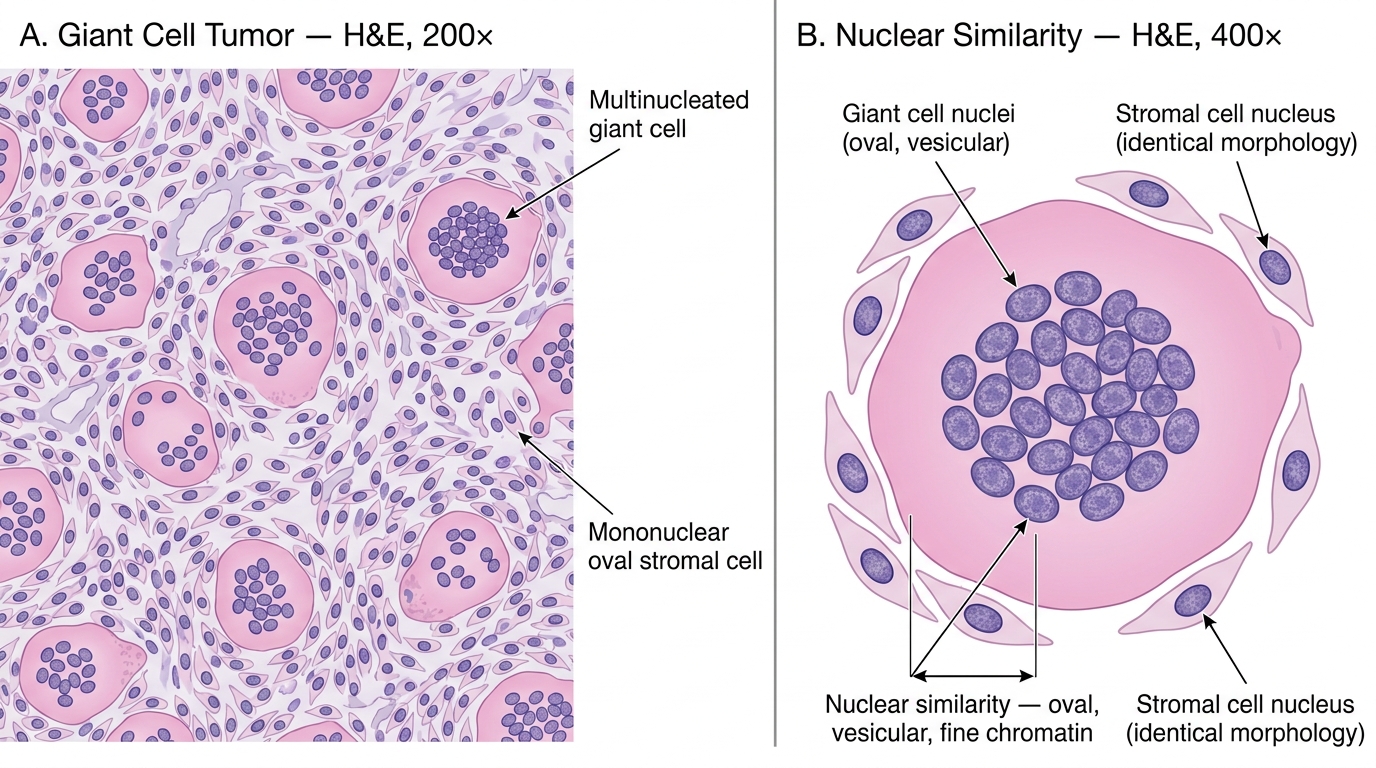
Giant Cell Tumor of Bone — H&E Histology: Even Giant Cell Distribution and Diagnostic Nuclear Similarity
Treatment: Curettage + bone graft (with high recurrence rate ~20–50%); denosumab (anti-RANKL antibody) for unresectable or recurrent GCT is now standard — it targets the osteoclast-like giant cells via the RANK-RANKL axis.
CLINICAL PEARL
Epiphysis vs metaphysis — the location rule that saves exam marks. Giant cell tumor almost exclusively involves the epiphysis of skeletally mature patients (fused growth plates). Osteosarcoma almost exclusively involves the metaphysis of skeletally immature patients (open growth plates). Ewing sarcoma involves the diaphysis of children. If an exam question gives you the site, you can narrow to one or two diagnoses before reading the histology. Memorize: Epiphysis → GCT; Metaphysis → osteosarcoma or chondrosarcoma; Diaphysis → Ewing.
Ewing Sarcoma
Ewing sarcoma is an aggressive bone tumor of childhood and adolescence with a defining chromosomal translocation and a characteristic radiologic appearance.
Key facts
- Age: 5–20 years (children and adolescents); rare over 30.
- Site: diaphysis (shaft) of long bones — femur, tibia, fibula, humerus. Also flat bones: pelvis, scapula, ribs.
- Male > female (1.5:1).
- Almost exclusively in Caucasian populations; rare in Africans and South Asians — an epidemiologic clue sometimes tested.
Etiology and pathogenesis
- Defining molecular event: t(11;22)(q24;q12) translocation in ~85% of cases, resulting in the EWSR1-FLI1 fusion gene.
- EWSR1-FLI1 encodes an aberrant transcription factor that drives uncontrolled proliferation of the precursor cells (neural crest/mesenchymal origin debated).
- Remaining 15%: other EWSR1 fusions (EWSR1-ERG most common).
Clinical features
- Localized pain and swelling, often intermittent initially.
- Systemic features: fever, elevated ESR, leukocytosis — mimics osteomyelitis. This is a classic clinical trap: Ewing sarcoma can mimic infection. Biopsy is essential when the clinical picture is ambiguous.
- Pathological fracture uncommon.
Radiologic features
- Diaphyseal, permeative lytic lesion (ill-defined, "moth-eaten" pattern).
- Onion-skin (laminated) periosteal reaction: successive layers of periosteal new bone deposited in concentric rings around the diaphysis, produced by the periosteum's reaction to repeated tumor expansion. This is the classical radiologic sign.
- Codman triangle may also appear in very aggressive cases.
- MRI shows extensive marrow infiltration and a large soft-tissue mass disproportionate to the bony destruction.
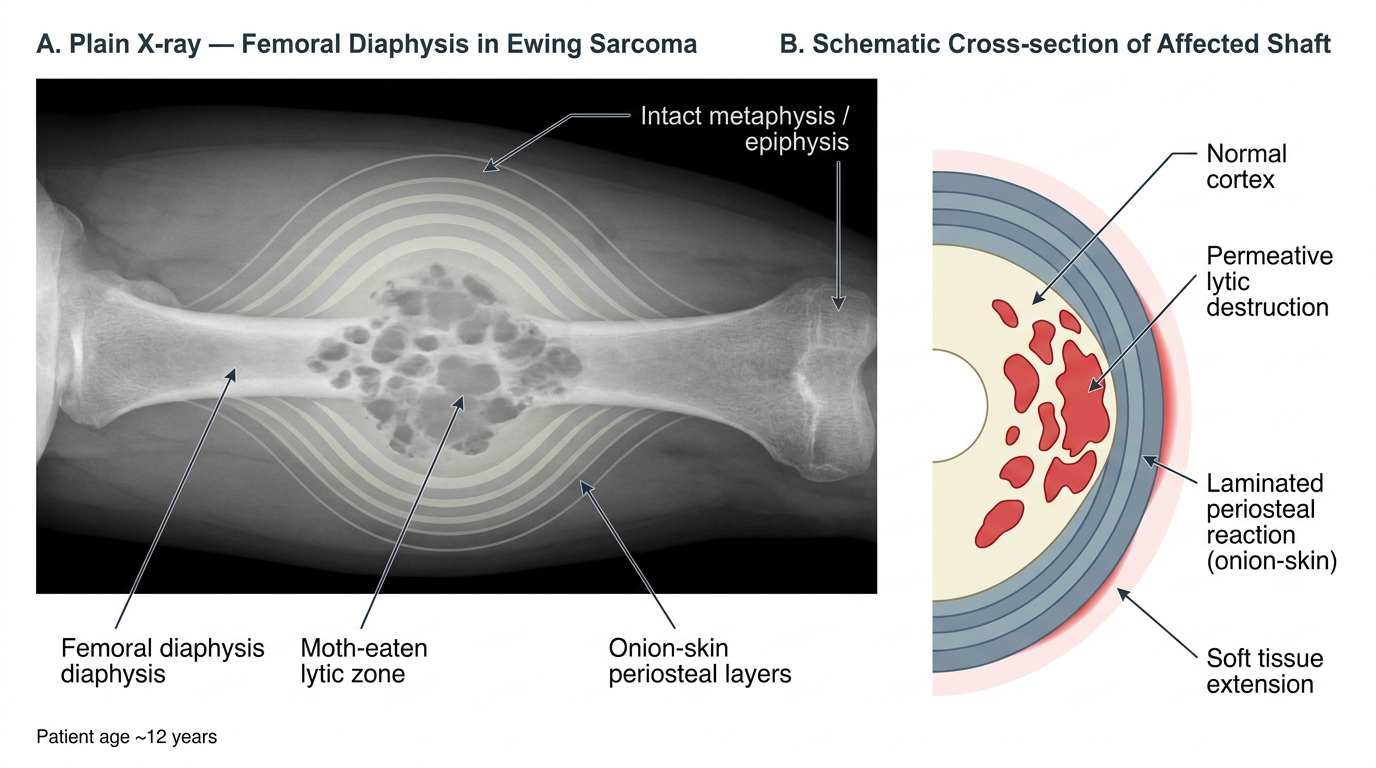
Plain X-ray and Schematic of Ewing Sarcoma — Femoral Diaphysis (Onion-Skin Periosteal Reaction)
Histology (DEFINITIVE)
- Sheets and nests of small, round, blue cells — monotonous, closely packed, with scanty cytoplasm, round nuclei, finely granular chromatin, and inconspicuous nucleoli.
- Glycogen-rich cytoplasm (PAS-positive vacuoles) — useful on frozen section.
- "Small round blue cell tumor": differential includes Ewing sarcoma, neuroblastoma, lymphoma (Burkitt's), rhabdomyosarcoma, and medulloblastoma. Immunohistochemistry distinguishes them: Ewing sarcoma is CD99 positive (diffuse membranous staining), FLI1 positive; FISH for EWSR1 rearrangement confirms.
- Homer Wright rosettes may be seen (pseudo-rosettes, not true rosettes).
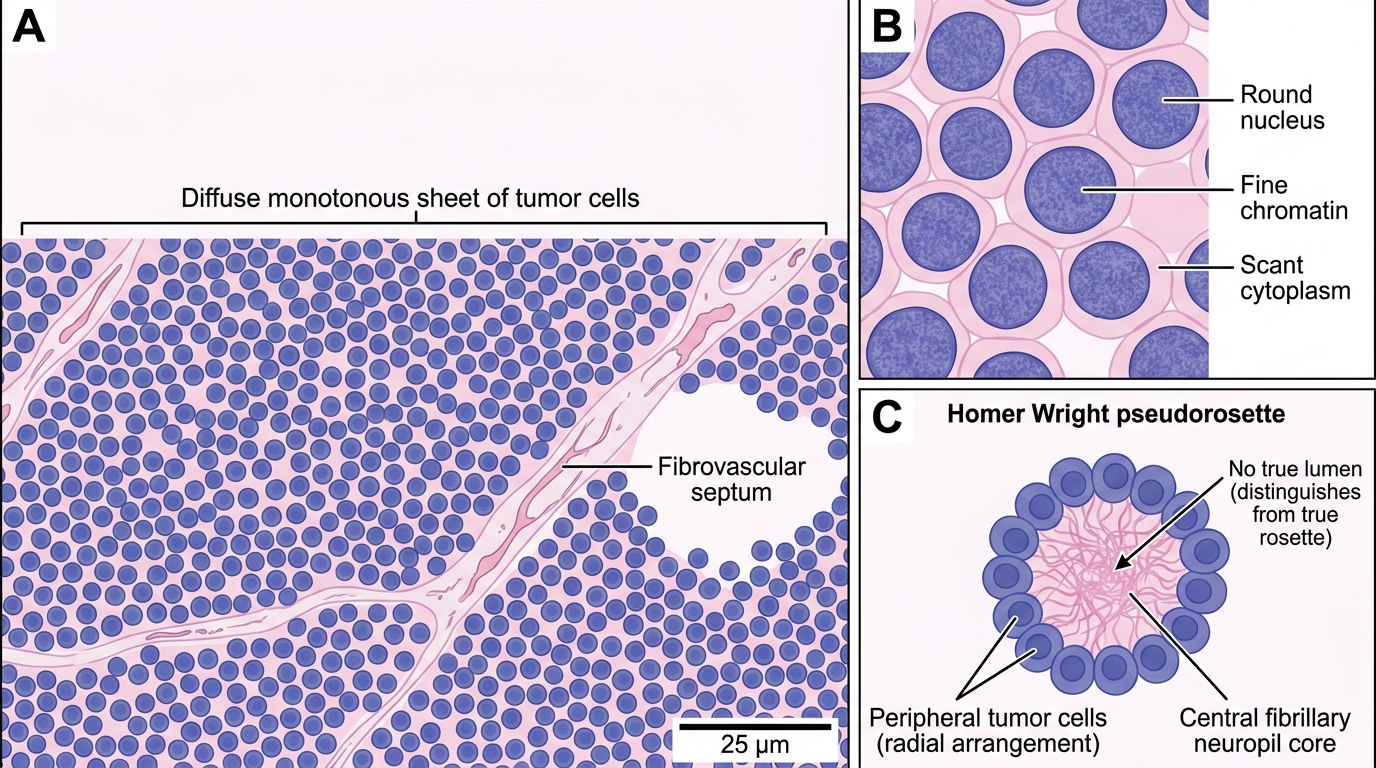
Histology of Ewing Sarcoma (H&E, 400×): Small Round Blue Cell Pattern
Spread and prognosis
- Hematogenous spread to lungs and bones.
- With multiagent chemotherapy + surgery ± radiation: 5-year survival ~70% for localized disease; ~30% for metastatic disease.
SELF-CHECK
A 12-year-old girl presents with 6 weeks of right thigh pain and fever. X-ray shows a diaphyseal lytic lesion with multiple concentric periosteal layers. Biopsy reveals sheets of small, round, blue cells with glycogen-rich cytoplasm. Which chromosomal translocation is the hallmark of this diagnosis?
A. t(9;22) BCR-ABL
B. t(14;18) BCL2-IGH
C. t(11;22) EWSR1-FLI1
D. t(8;14) MYC-IGH
Reveal Answer
Answer: C. t(11;22) EWSR1-FLI1
The combination of (1) a child, (2) diaphyseal location, (3) onion-skin periosteal reaction, and (4) small round blue cells = Ewing sarcoma, defined by t(11;22)(q24;q12) — EWSR1-FLI1 fusion in ~85% of cases. t(9;22) is chronic myeloid leukemia; t(14;18) is follicular lymphoma; t(8;14) is Burkitt's lymphoma.