Page 1 of 34
PA26.1-2 | Atherosclerosis & Aneurysms — SDL Guide
Learning Objectives
- Distinguish arteriosclerosis from atherosclerosis and classify its subtypes
- Explain the response-to-injury hypothesis of atherogenesis step by step
- Describe the morphology and distribution of the atheromatous plaque
- List the modifiable and non-modifiable risk factors for atherosclerosis
- Enumerate the complications of atherosclerosis including plaque rupture and thrombosis
- Define true vs false aneurysm and classify aneurysms by shape and aetiology
- Apply Laplace's Law to explain aneurysm enlargement and rupture
- Describe the pathology and complications of abdominal aortic aneurysm (AAA) and aortic dissection
INSTRUCTIONS
Arterial disease kills more Indians than any other disease category — ischaemic heart disease, stroke, and aortic catastrophes all trace back to the wall of a blood vessel. Mastering atherosclerosis and aneurysms is therefore not optional pathology; it is the mechanistic backbone of clinical medicine you will apply every day as a physician. This module builds that foundation through the response-to-injury hypothesis, plaque biology, and vascular mechanics — concepts that recur in Pharmacology, Medicine, Surgery, and Cardiology.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch 11 (Blood Vessels) (textbook)
- Harsh Mohan — Textbook of Pathology, 8th ed., Ch 15 (Blood Vessels) (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 62-year-old man with a 30-year smoking history and poorly controlled hypertension presents to the emergency department with sudden, tearing chest pain radiating to the back. His blood pressure differs by 40 mmHg between his two arms. A CT angiogram reveals a Stanford Type A aortic dissection. Four hours earlier, a 58-year-old woman was brought in pulseless after a ruptured abdominal aortic aneurysm found incidentally on screening ultrasound six months ago — her family had declined surgery.
These are not rare outliers. They are the endpoint of decades-long arterial disease that started silently with a fatty streak in adolescence. Understanding why and how arteries fail is the mission of this module.
WHY THIS MATTERS
- PA26.1 maps directly to the MCI/NBE long-case format: examiners ask for risk stratification, pathogenesis mechanism, and morphology description of plaques.
- PA26.2 is a high-yield surgery-pathology crossover: AAA, dissecting aneurysm, and berry aneurysm appear repeatedly in USMLE-style integrated questions.
- Clinico-pathological correlation: every MI, stroke, claudication, and renal artery stenosis you manage in wards has its origin in concepts you will learn today.
- Year-1 foundation used here: AN vascular anatomy (aorta, circle of Willis), PY endothelial physiology, BI lipid metabolism.
RECALL
Before proceeding, mentally revisit:
1. The three layers of an arterial wall — tunica intima, media, adventitia — and the cell types in each.
2. The general structure of a lipoprotein: which lipoprotein carries cholesterol to peripheral tissues (LDL) and which carries it back to the liver (HDL)?
3. From Physiology: what is endothelial-derived nitric oxide (eNO) and why is its loss pro-thrombotic?
4. From Biochemistry: what is oxidised LDL (ox-LDL) and why is it more atherogenic than native LDL?
If any of these feel uncertain, spend two minutes with your Year-1 notes before continuing — this module builds on all four.
Arteriosclerosis: The Umbrella Term
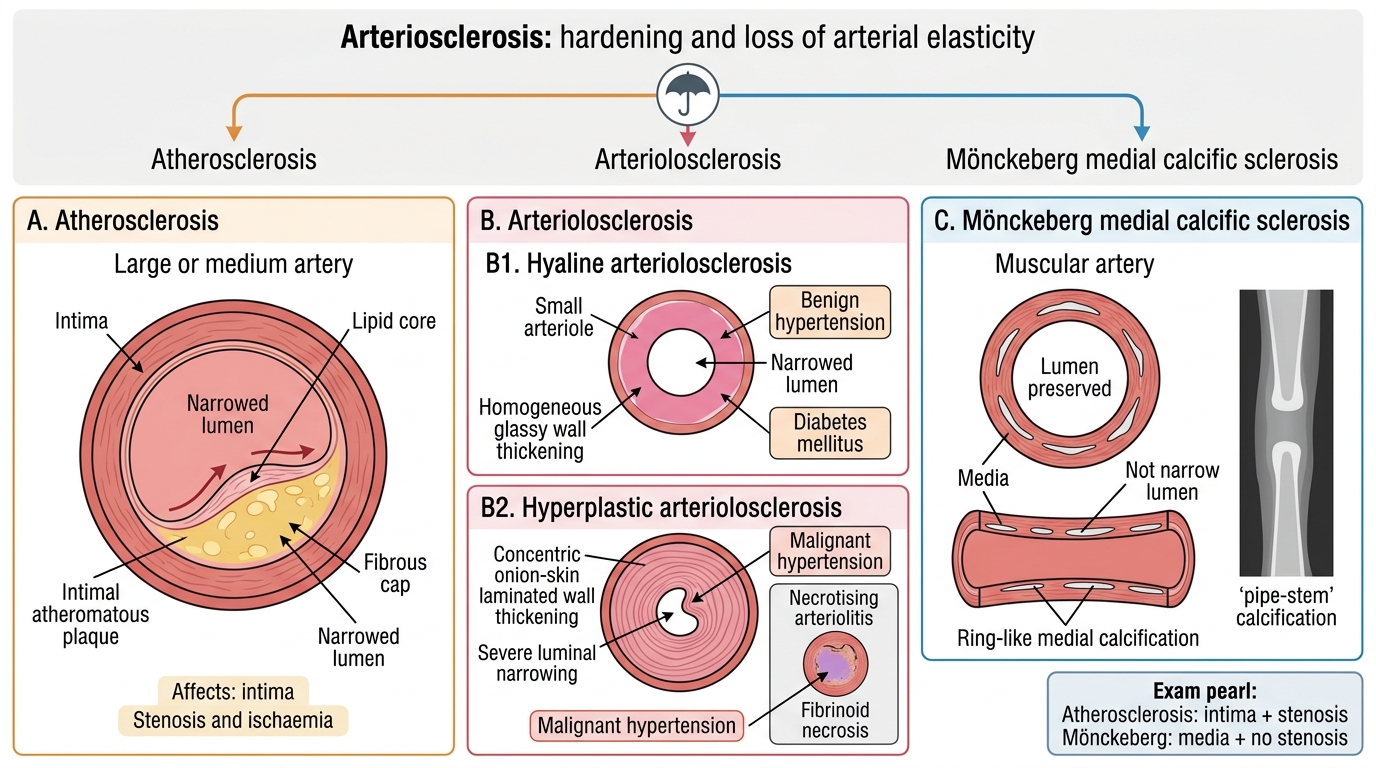
Arteriosclerosis: Major Forms and Exam Distinctions
Arteriosclerosis (literally 'hardening of arteries') is a generic term for thickening and loss of elasticity of the arterial wall. Three distinct processes fall under this umbrella:
1. Atherosclerosis — the dominant and most clinically important form; involves large and medium elastic/muscular arteries; driven by lipid deposition and inflammation. (Covered in depth in the next sections.)
2. Arteriolosclerosis — affects small arteries and arterioles; two morphological patterns:
• Hyaline arteriolosclerosis: pink, homogeneous, glassy thickening of the vessel wall due to plasma protein leakage; associated with benign hypertension and diabetes mellitus (hyperglycaemia accelerates protein glycation and basement membrane thickening).
• Hyperplastic arteriolosclerosis (onion-skin lesion): concentric laminated thickening of the wall with luminal narrowing; seen in malignant hypertension (diastolic BP >120 mmHg). Can progress to necrotising arteriolitis with fibrinoid necrosis.
3. Mönckeberg medial calcific sclerosis — dystrophic calcification of the media of muscular arteries (typically femoral, tibial, radial); ring-like calcifications visible on X-ray ('pipe-stem' arteries); does NOT narrow the lumen and is NOT atheromatous; more common in elderly and diabetics; usually an incidental finding.
Key distinction for exams: Mönckeberg affects media, does not cause stenosis; atherosclerosis affects intima, causes stenosis and ischaemia.
Atherosclerosis: Risk Factors
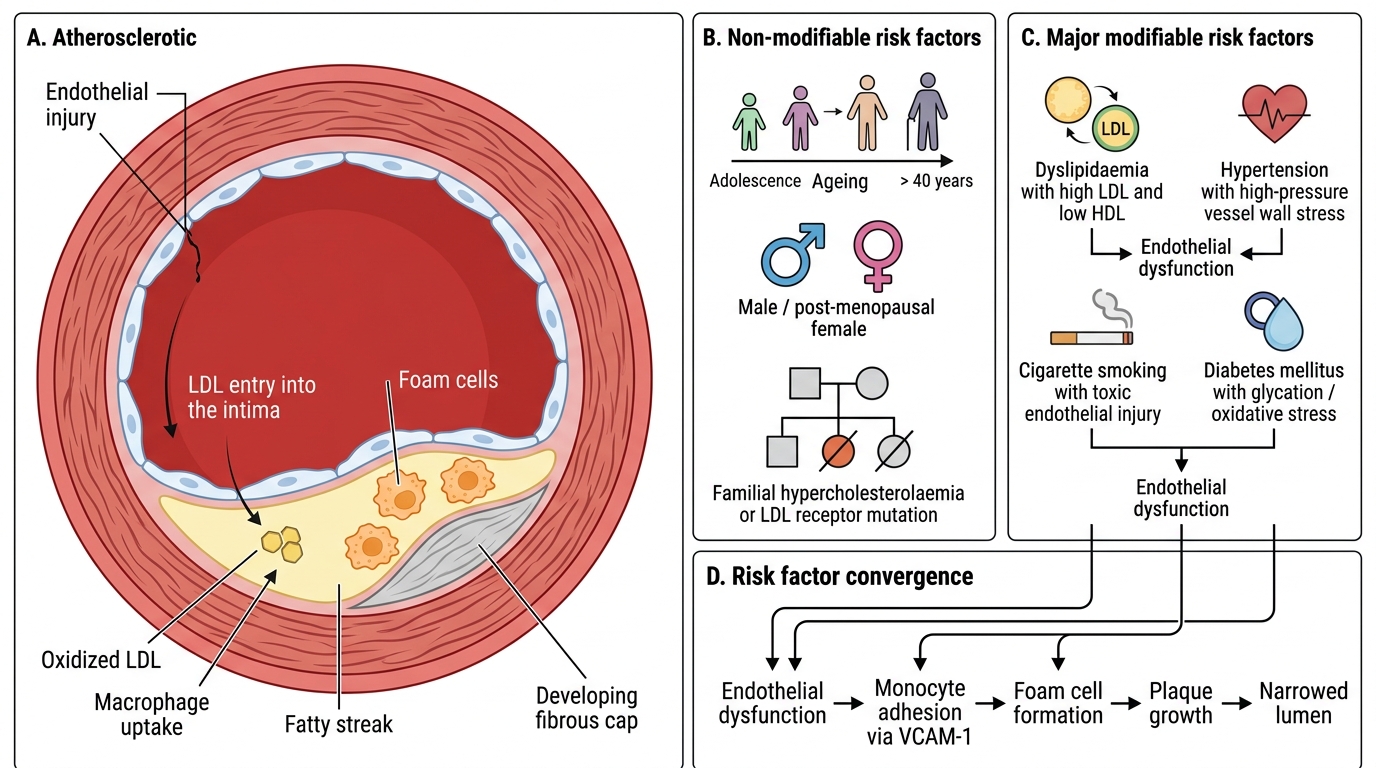
Atherosclerosis: Risk Factors and Pathogenesis
Atherosclerosis is a multifactorial disease. Risk factors are classically divided:
Non-modifiable risk factors:
| Factor | Comment |
|---|---|
| Age | Risk rises progressively; lesions begin in adolescence but manifest clinically after 40 |
| Sex | Men at higher risk at all ages; women 'catch up' post-menopause (loss of oestrogen's protective effect on lipid profile and endothelium) |
| Genetics / family history | Familial hypercholesterolaemia (FH): LDL-receptor mutations → severe premature atherosclerosis |
Modifiable risk factors (MAJOR):
| Factor | Mechanism |
|---|---|
| Dyslipidaemia (↑LDL, ↓HDL) | LDL enters intima, oxidised → foam cell formation; HDL exports cholesterol, is protective |
| Hypertension | Haemodynamic injury to endothelium; increases LDL uptake; upregulates VCAM-1 |
| Cigarette smoking | Oxidative stress; CO damages endothelium; ↑LDL, ↓HDL; promotes thrombosis |
| Diabetes mellitus | Glycation of LDL (harder for receptors to clear); endothelial dysfunction; accelerates foam-cell conversion |
Other contributing factors: obesity (central/android), physical inactivity, C-reactive protein (marker of inflammation), homocysteinaemia, lipoprotein(a), thrombogenic risk factors.
> Clinical pearl: The Framingham risk score integrates age, sex, LDL, HDL, BP, and smoking to estimate 10-year cardiovascular risk — a direct clinical translation of these pathology risk factors.
SELF-CHECK
A 45-year-old woman has total cholesterol 280 mg/dL, LDL 200 mg/dL, HDL 35 mg/dL, BP 155/95 mmHg, and smokes 20 cigarettes/day. Which single intervention would have the GREATEST impact on reducing her atherosclerotic risk?
A. Start a high-intensity statin to lower LDL
B. Begin antihypertensive therapy
C. Advise smoking cessation
D. Prescribe niacin to raise HDL
Reveal Answer
Answer: C. Advise smoking cessation
Smoking cessation (option C) addresses multiple simultaneous mechanisms: it eliminates oxidative endothelial injury, reduces CO-mediated damage, reverses the LDL↑/HDL↓ dyslipidaemia induced by smoking, and dramatically reduces thrombotic risk — often achieving larger net risk reduction than any single pharmacological agent. Statins and antihypertensives are important adjuncts, but cessation is the highest-yield single step in a heavy smoker.
Pathogenesis of Atherosclerosis: Response-to-Injury Hypothesis
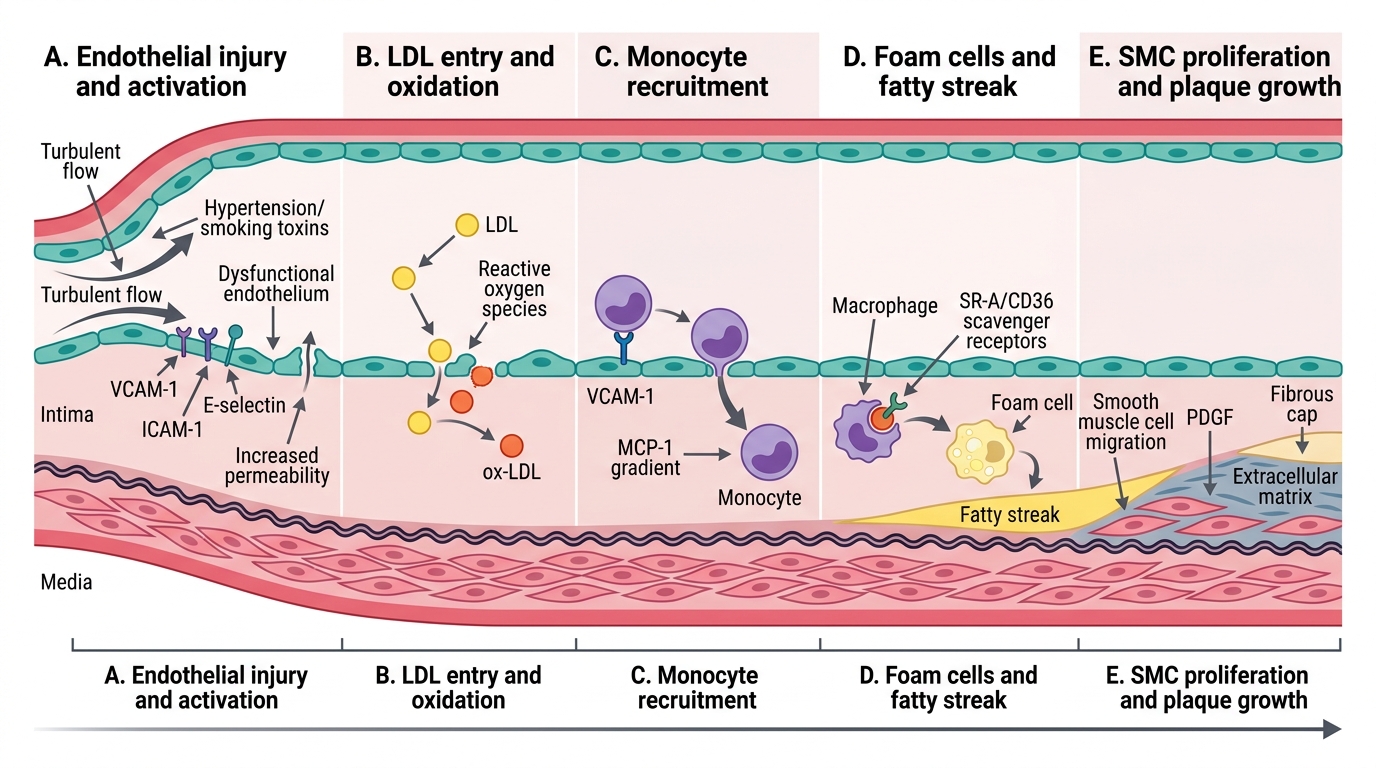
Response-to-Injury Hypothesis of Atherosclerosis
The most accepted mechanistic framework is the response-to-injury hypothesis (Ross, 1990s), which unifies lipid, inflammatory, and haemodynamic inputs into a single sequence:
Step 1 — Endothelial injury and dysfunction
Triggers: haemodynamic turbulence (branch points, bifurcations), ox-LDL, hypertension, smoking toxins, homocysteine. The endothelium loses its anti-thrombotic, anti-inflammatory state — it expresses adhesion molecules (VCAM-1, ICAM-1, E-selectin) and becomes permeable.
Step 2 — LDL entry and oxidation
LDL accumulates in the intima. Local reactive oxygen species (from macrophages, smoking) oxidise it to ox-LDL. Ox-LDL is proinflammatory and cannot be cleared by the normal LDL receptor.
Step 3 — Monocyte recruitment and foam cell formation
Circulating monocytes adhere to activated endothelium (via VCAM-1/MCP-1 gradient) → migrate into intima → differentiate into macrophages → engulf ox-LDL via scavenger receptors (SR-A, CD36) → become foam cells (lipid-laden macrophages) → aggregate into fatty streaks (flat, yellow intimal deposits; seen from adolescence; reversible).
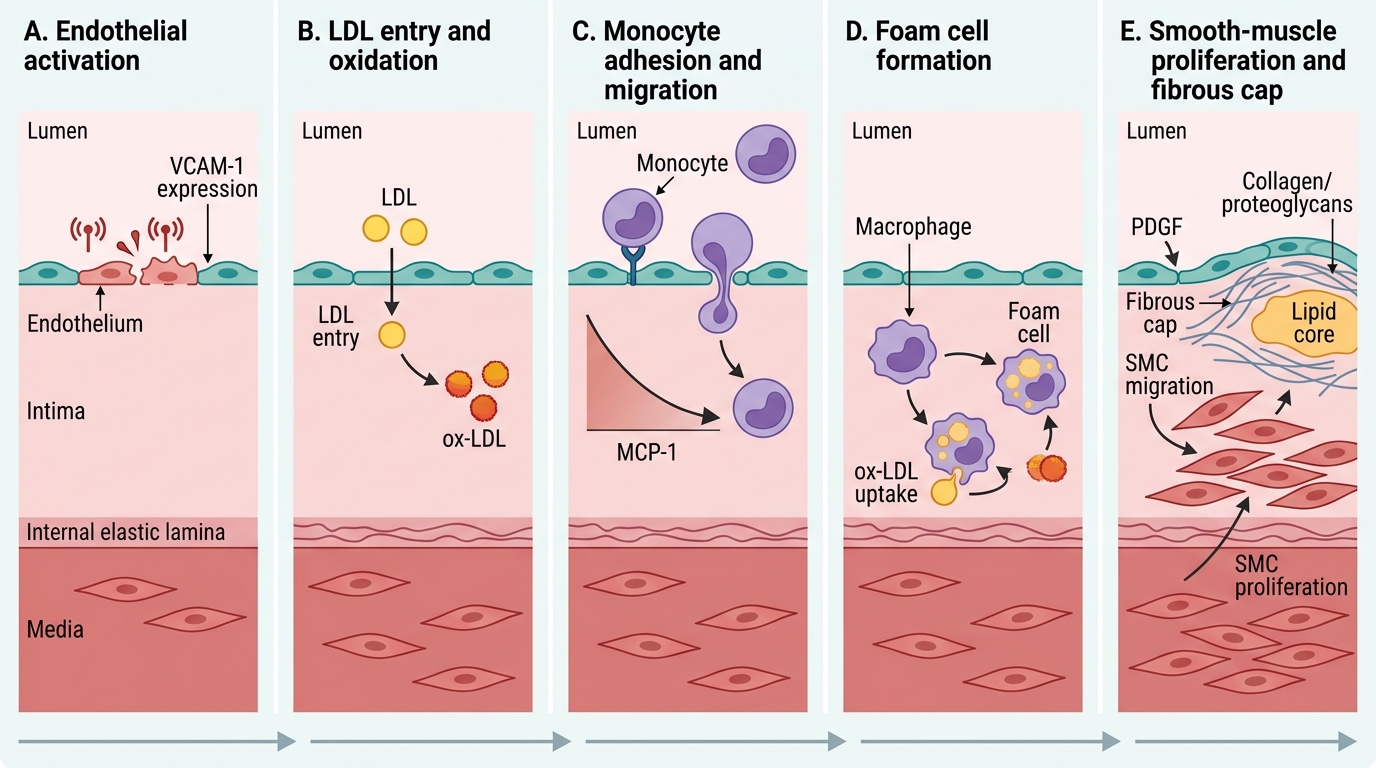
Response-to-Injury Sequence in Atherosclerosis
Step 4 — Smooth muscle cell (SMC) migration and proliferation
Activated macrophages and platelets release PDGF (platelet-derived growth factor) and FGF → stimulate medial SMCs to migrate into the intima and proliferate. SMCs synthesise extracellular matrix (collagen, proteoglycans) → fibrous cap forms over the lipid core.
Step 5 — Progressive plaque formation
Continued lipid accumulation, SMC proliferation, collagen deposition, and inflammation → fibrous atheromatous plaque (described below). Lesion grows eccentrically into the lumen.