Page 11 of 34
PA26.3 | Heart Failure — SDL Guide
Learning Objectives
- Define heart failure and explain why the heart fails to meet metabolic demand
- Describe the three major compensatory mechanisms (Frank-Starling, neurohormonal, hypertrophy/remodelling) and explain how each eventually maladapts
- Distinguish left from right heart failure by cause, haemodynamic consequence, and pathological findings — including pulmonary oedema, heart failure cells, and nutmeg liver
- Differentiate systolic (reduced-EF) from diastolic (preserved-EF) heart failure, and high-output from low-output failure
- List the major complications of heart failure and describe the role of BNP as a biomarker
- Recall NYHA functional classes and ACC/AHA stages in clinical context
INSTRUCTIONS
Heart failure is the final common pathway of almost every cardiac disease you will encounter in medicine. Understanding why the heart fails — and why its own attempts to compensate ultimately worsen the outcome — is the foundation of all rational management. This module builds clinico-pathological fluency: structure → dysfunction → bedside findings. Work through each section in order; the micro-quizzes are calibrated to the level of your final exam.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch 12 — The Heart (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch 16 — Heart Failure (textbook)
- Kumar & Clark's Clinical Medicine, 10th ed., Ch 18 — Cardiovascular Disease (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 58-year-old man with long-standing hypertension presents breathless at rest. His chest X-ray shows cardiomegaly with bilateral basal haziness. His jugular veins are distended at 45°, both ankles are pitted to mid-calf, and there is a tender hepatomegaly. His ECG shows left ventricular hypertrophy. His BNP is 1,200 pg/mL.
His heart has been compensating for years — and has now run out of tricks.
By the end of this module you will be able to read that presentation as a physiological story: from the initial insult, through each compensatory mechanism, to the moment the compensation becomes the disease.
WHY THIS MATTERS
Heart failure affects more than 26 million people worldwide and is the leading cause of hospitalisation in adults over 65. In India, hypertensive and rheumatic heart disease drive a disproportionate burden. For a CBME-trained physician, understanding heart failure is not optional — it underlies the management of IHD, CKD, liver congestion, pulmonary hypertension, and anaemia all at once. PA26.3 maps directly to clinical postings where you will encounter acute decompensated heart failure on the wards.
RECALL
Before reading on, take 90 seconds to recall:
- What is cardiac output? How is it determined by preload, afterload, and contractility?
- What does the Frank-Starling law state about fibre length and force of contraction?
- Name two hormones of the renin-angiotensin-aldosterone system (RAAS).
- Where does lymphatic drainage of the lung go, and what happens when it is overwhelmed?
If any of these feel uncertain, revisit your Year-1 Physiology notes on cardiac mechanics (PY8) before proceeding — this module builds directly on them.
Definition and Core Concept
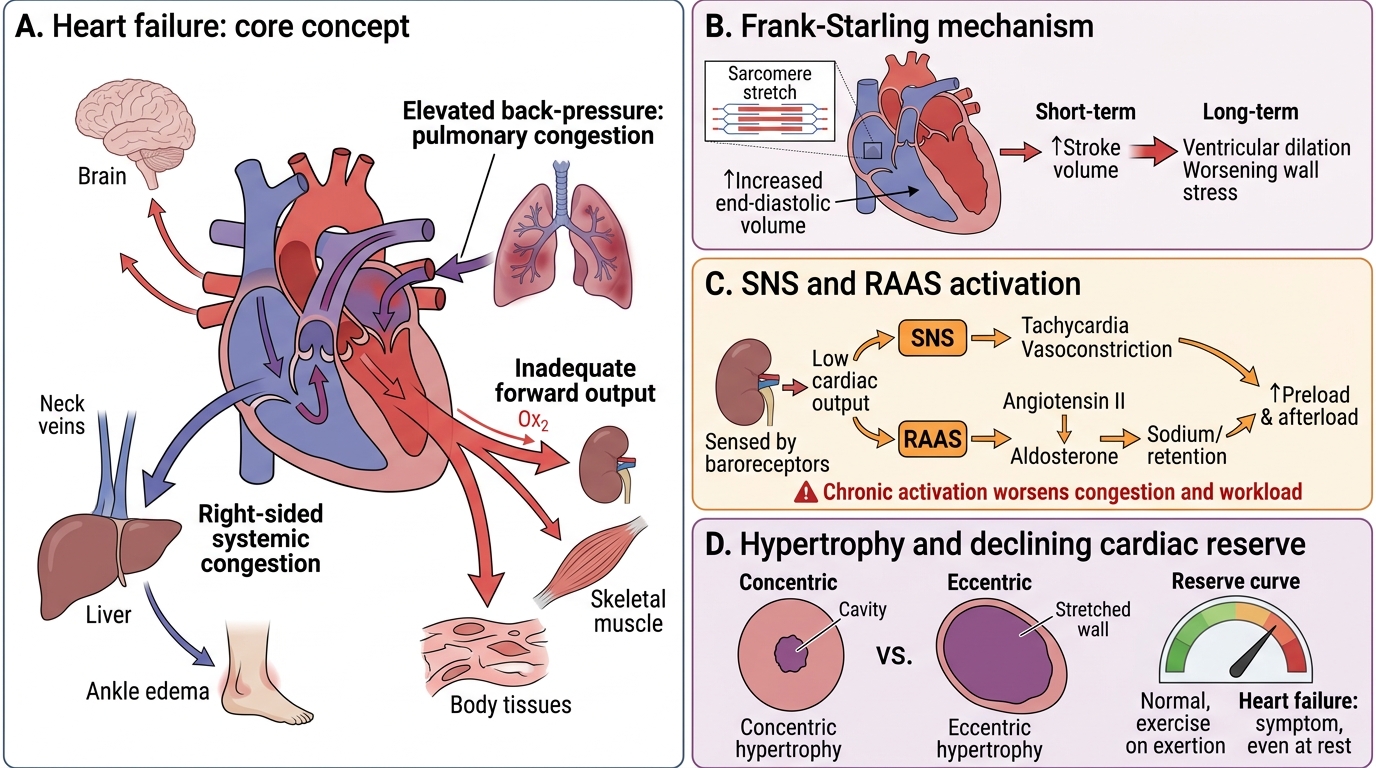
Heart Failure: Core Concept and Compensatory Mechanisms
Heart failure (HF) is a clinical syndrome in which the heart is unable to pump sufficient blood to meet the metabolic demands of the body's tissues, or can do so only at the cost of abnormally elevated filling pressures.
Two points are essential in this definition:
- Inadequate forward output — organs receive too little oxygenated blood (low-output state).
- Elevated back-pressure — blood dams up behind the failing ventricle, congesting the lungs (left failure) or systemic circulation (right failure).
In early failure, the heart deploys compensatory mechanisms that temporarily restore output. Over months to years these compensations become maladaptive — they worsen the very problem they were recruited to solve. This paradox is the central lesson of heart failure pathophysiology.
Cardiac reserve is the ability to increase output during stress (exercise, fever, anaemia). Heart failure erodes this reserve; the patient first notices symptoms only on exertion, then at rest.
Compensatory Mechanisms — and Their Maladaptation
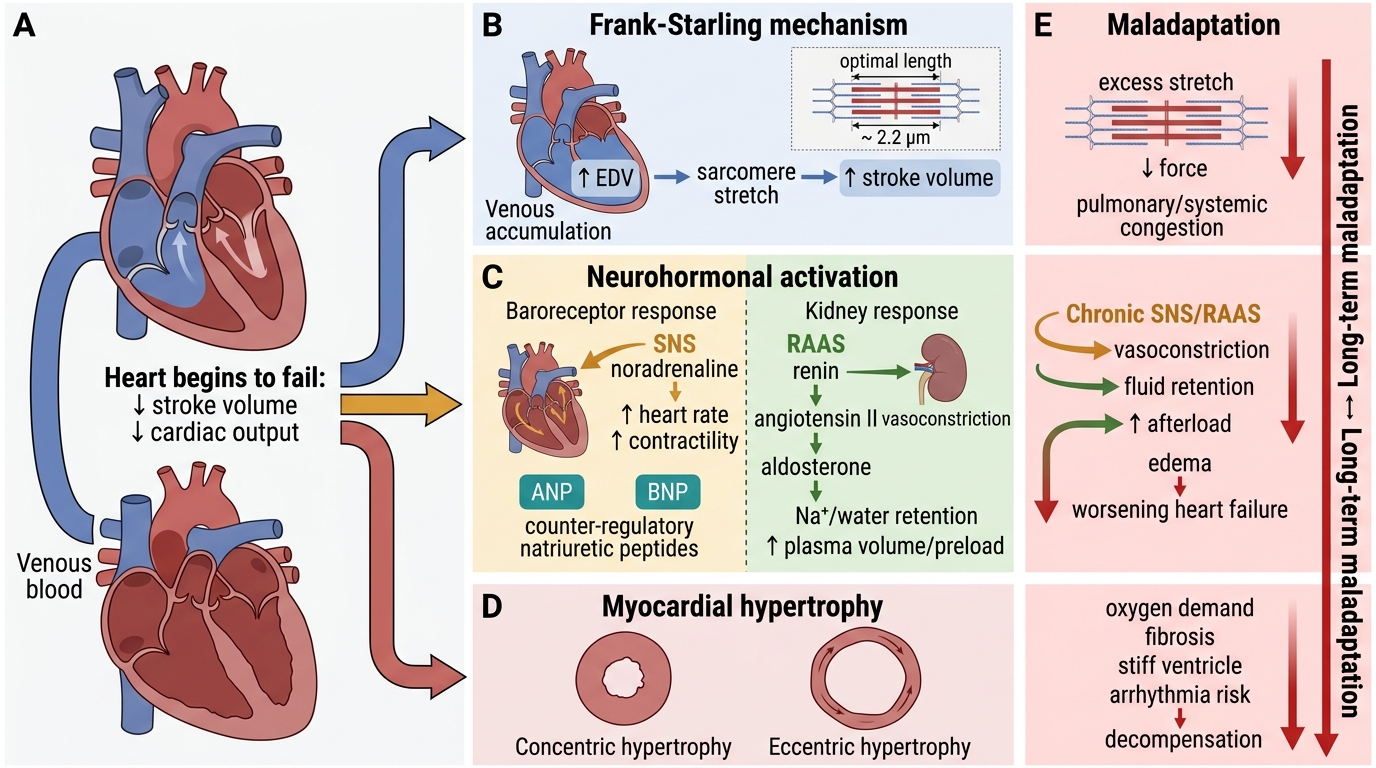
Compensatory Mechanisms in Heart Failure
Three mechanisms are deployed when the heart begins to fail. Each works in the short term; each eventually harms in the long term.
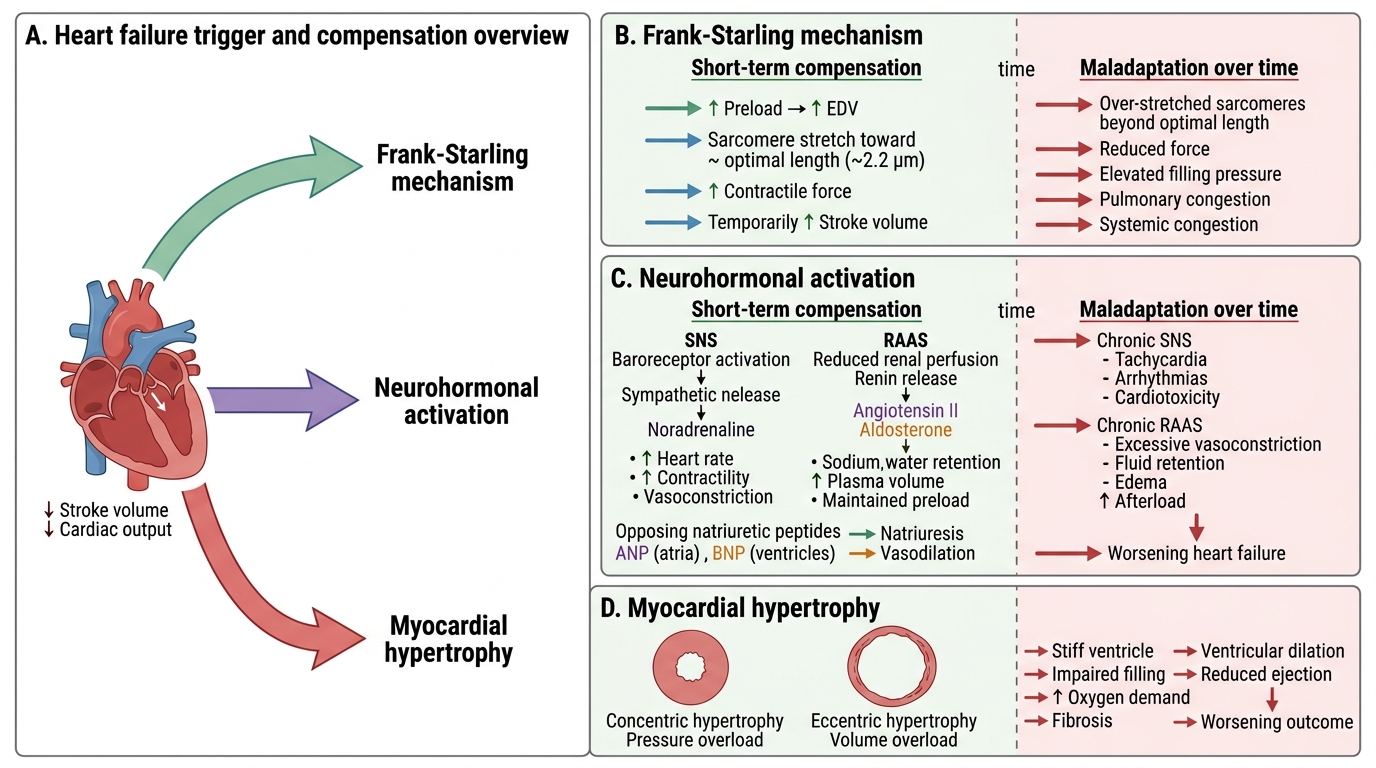
Compensatory Mechanisms in Heart Failure
1. Frank-Starling mechanism
When stroke volume falls, venous blood accumulates → end-diastolic volume (EDV) rises → sarcomere stretch increases → contractile force increases → stroke volume is temporarily restored. This is the Frank-Starling law at work. Maladaptation: beyond optimal sarcomere length (~2.2 µm), further stretch reduces force. Chronically elevated filling pressures cause pulmonary and systemic congestion.
2. Neurohormonal activation
Falling cardiac output activates baroreceptors → sympathetic nervous system (SNS) releases noradrenaline → heart rate and contractility rise. Simultaneously, reduced renal perfusion activates the renin-angiotensin-aldosterone system (RAAS): angiotensin II causes vasoconstriction and aldosterone causes Na⁺/water retention, expanding plasma volume to maintain preload. Natriuretic peptides (ANP from atria, BNP from ventricles) are released in response to wall stretch and oppose the RAAS — promoting natriuresis and vasodilation. Maladaptation: chronic SNS activation causes tachycardia, arrhythmias, and direct cardiotoxicity; chronic RAAS activation causes afterload increase and myocardial fibrosis; natriuretic peptides become insufficient against sustained RAAS drive.
3. Myocardial hypertrophy and remodelling
Persistent haemodynamic overload drives myocardial hypertrophy — an increase in cardiomyocyte size (not number). The pattern depends on the type of overload:
- Concentric hypertrophy: pressure overload (hypertension, aortic stenosis) → sarcomeres added in parallel → wall thickens, cavity remains normal or small. Useful short-term; diastolic stiffness is the long-term price.
- Eccentric hypertrophy: volume overload (regurgitant valves, dilated cardiomyopathy) → sarcomeres added in series → wall thins, cavity dilates. Wall stress rises (LaPlace: T = Pr/2h), further impairing systolic function.
Remodelling refers to the progressive geometric changes in chamber shape, including cardiomyocyte loss by apoptosis and necrosis, interstitial fibrosis (activated by angiotensin II and TGF-β), and chamber dilation. Once established, remodelling is self-perpetuating — it is the pathological substrate of "end-stage heart failure."
SELF-CHECK
A patient with aortic stenosis develops left ventricular hypertrophy. Sarcomeres are added in parallel, and wall thickness increases without significant cavity enlargement. This pattern is best described as:
A. Eccentric hypertrophy due to volume overload
B. Concentric hypertrophy due to pressure overload
C. Physiological hypertrophy seen in athletes
D. Dilated cardiomyopathy pattern
Reveal Answer
Answer: B. Concentric hypertrophy due to pressure overload
Pressure overload (e.g. aortic stenosis, hypertension) drives sarcomere addition in parallel, producing concentric hypertrophy: thickened wall, normal or reduced cavity. Volume overload (regurgitation) adds sarcomeres in series, producing eccentric hypertrophy with cavity dilation. Athlete's heart is a physiological form of concentric or mildly eccentric hypertrophy and is reversible.
Classification of Heart Failure
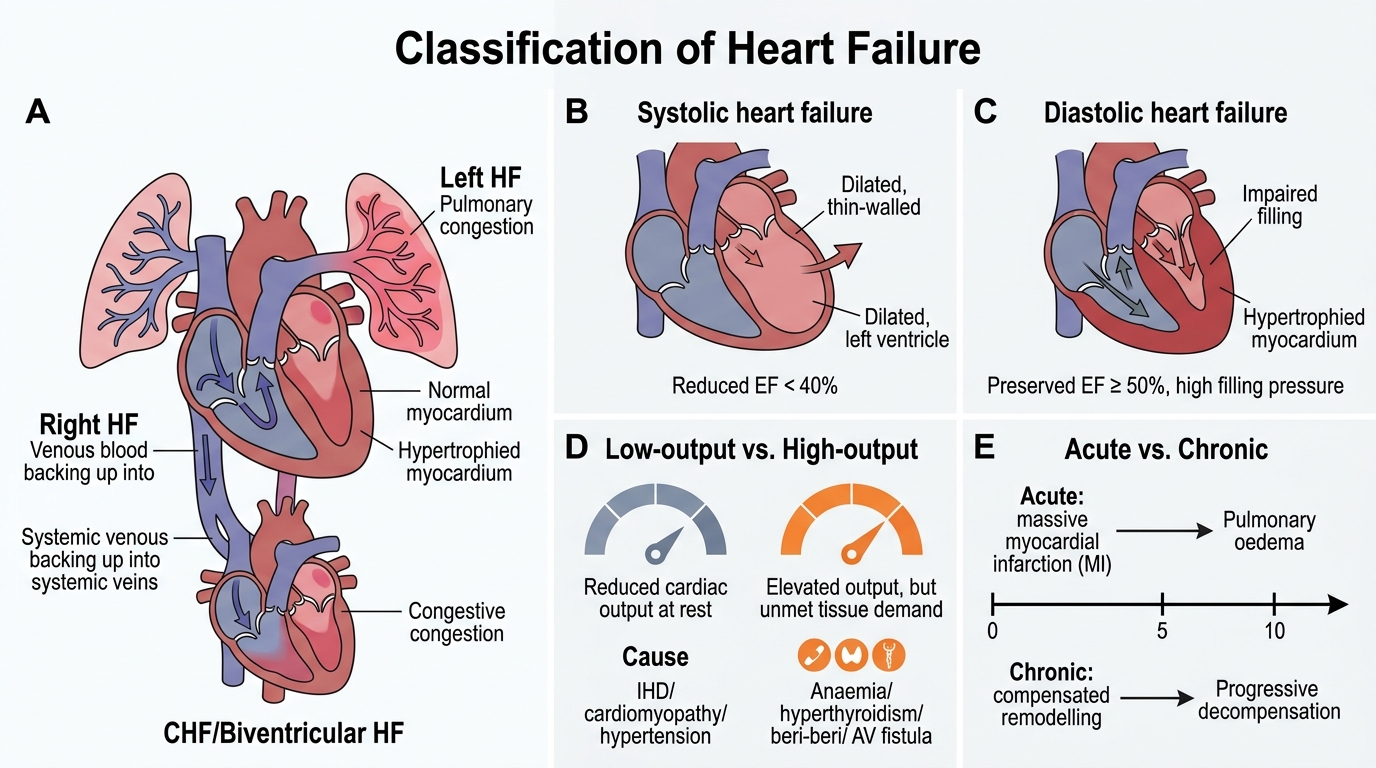
Classification of Heart Failure
Heart failure is classified along several axes — each carries distinct clinical and pathological implications.
Left vs Right vs Biventricular
The most important anatomical distinction: left HF dams blood into the pulmonary circulation; right HF dams blood into the systemic venous circulation. Congestive heart failure (CHF) implies both circuits are congested — almost always because untreated left HF eventually overloads the right ventricle.
Systolic vs Diastolic
- Systolic (reduced EF) heart failure: impaired contractility → ejection fraction (EF) < 40%. Common causes: IHD, dilated cardiomyopathy. The heart dilates and thins.
- Diastolic (preserved EF) heart failure: impaired ventricular relaxation and compliance → EF ≥ 50% but filling pressures are elevated. Common causes: hypertension, hypertrophic cardiomyopathy, constrictive pericarditis. Wall is thickened and stiff — the heart cannot fill, not because it won't pump.
High-output vs Low-output
- Low-output HF (the majority): cardiac output is subnormal at rest. IHD, cardiomyopathy, hypertension.
- High-output HF: heart fails to keep up with supranormal metabolic demand despite initially elevated output. Causes: severe anaemia, hyperthyroidism, beri-beri (thiamine deficiency), arteriovenous fistulae. Paradox: output appears high but is relatively insufficient.
Acute vs Chronic
Acute (sudden onset — e.g., massive MI) allows no time for compensatory remodelling; pulmonary oedema or cardiogenic shock can develop within minutes to hours. Chronic failure allows weeks/months of adaptive changes; the patient presents with the gradual symptoms described in the next sections.
SELF-CHECK
A 70-year-old woman with known hypertension presents with exertional dyspnoea. Echocardiography shows a non-dilated, concentrically hypertrophied left ventricle with EF of 62%. This is most consistent with:
A. Systolic heart failure (HFrEF)
B. Diastolic heart failure (HFpEF)
C. High-output heart failure
D. Right heart failure
Reveal Answer
Answer: B. Diastolic heart failure (HFpEF)
Preserved EF (≥ 50%) with elevated filling pressures (dyspnoea on exertion) in a hypertensive patient with LV hypertrophy is classic HFpEF (heart failure with preserved ejection fraction = diastolic heart failure). The stiff, non-compliant ventricle cannot fill adequately. HFrEF has EF < 40% with a dilated, thin-walled ventricle.