Page 15 of 26
PA31.{5,7} | Diabetes Mellitus & Pancreatic Cancer — SDL Guide
Learning Objectives
- Classify diabetes mellitus into Type 1, Type 2, MODY, secondary, and gestational subtypes and compare their etiopathogenesis
- Describe the immunological and genetic mechanisms underlying Type 1 DM including HLA associations and insulitis
- Explain the pathophysiology of insulin resistance and progressive β-cell failure in Type 2 DM in the context of obesity and metabolic syndrome
- Apply the ADA diagnostic laboratory criteria for diabetes: fasting glucose, random glucose, HbA1c, and OGTT thresholds
- Distinguish diabetic ketoacidosis (DKA) from hyperosmolar hyperglycemic state (HHS) on pathophysiology, biochemistry, and clinical grounds
- Describe the morphological and pathological basis of diabetic complications — microangiopathy, nephropathy (Kimmelstiel-Wilson nodular glomerulosclerosis), retinopathy, neuropathy, macrovascular atherosclerosis, and diabetic foot
- Describe the etiology, molecular pathogenesis (KRAS, p16, TP53, SMAD4), precursor lesion (PanIN), gross and microscopic morphology, clinical manifestations, lab markers, and metastatic behaviour of pancreatic ductal adenocarcinoma
INSTRUCTIONS
Diabetes mellitus is India's most important non-communicable disease — 101 million Indians have diabetes (IDF Atlas 2021), and you will see its complications across every clinical posting, from the medical ward to the ophthalmology OPD. Pancreatic cancer is the lethal silent partner: it kills more than 90% of its patients within 5 years, largely because it presents late. Together, these two conditions map to 'disease of the endocrine pancreas' — a single cluster that bridges metabolic medicine and oncology. Work through this SDL carefully; the pathological mechanisms you learn here will sharpen your clinical reasoning for decades.
References
- Robbins & Cotran Pathologic Basis of Disease, 10th ed., Ch. 24 (The Endocrine System) + Ch. 19 (The Pancreas) (textbook)
- Harsh Mohan Textbook of Pathology, 8th ed., Ch. 28 (Endocrine Organs) (textbook)
- ADA Standards of Medical Care in Diabetes — 2024, Diabetes Care 47(Suppl 1) (guideline)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 14-year-old boy collapses in the school corridor. His friends say he had been complaining of excessive thirst and frequent urination for three weeks. On arrival at the emergency, he is drowsy, breathing in deep, rapid sighs, and his breath smells fruity. Blood glucose: 28 mmol/L. Blood gas: pH 7.12, bicarbonate 8 mEq/L. Ketones ++++ on urine dipstick.
Across town, a 52-year-old software engineer with a BMI of 32 kg/m² attends a routine health camp. His random blood glucose is 14.1 mmol/L (254 mg/dL). He has no symptoms. He is sent for a fasting glucose the next morning: 8.4 mmol/L. He is diagnosed with Type 2 diabetes — a disease he has probably had silently for years.
Same organ. Same molecule (insulin). Entirely different mechanisms. This SDL unpacks both — and adds the cancer that hides in the same gland until it is almost always too late.
WHY THIS MATTERS
Diabetes mellitus is directly tested in PA31.5 and is one of the highest-yield NMC CBUC competencies in systemic pathology. You will encounter its complications in Internal Medicine, Ophthalmology, Nephrology, Neurology, Vascular Surgery, and the ICU. Understanding the pathological basis of each complication — not just its name — is what distinguishes a pathologist-trained clinician from a protocol-follower.
Pancreatic ductal adenocarcinoma (PA31.7) is examined in the context of its dismal prognosis and why early detection remains so difficult. The molecular pathway (KRAS → p16 → TP53 → SMAD4) is a model for understanding oncogenesis in solid tumours generally. Both conditions appear in university examinations as long-answer questions, viva questions, and structured short-answer questions.
RECALL
Before continuing, activate what you already know:
- Where in the pancreas are the islets of Langerhans located, and what cell types do they contain? What does each cell type secrete?
- What is the difference between insulin-dependent and non-insulin-dependent glucose uptake in tissues?
- What is HbA1c, and why does it reflect glycaemic control over the preceding 8–12 weeks?
- What are the two major classes of autoimmune disease — organ-specific vs systemic — and which class does Type 1 DM belong to?
- What is apoptosis, and how does it differ from necrosis? (Relevant to β-cell loss in Type 1 DM.)
If any of these feel uncertain, flag them as you go — you will encounter each concept in the blocks ahead.
Normal Islet Anatomy and the Insulin Axis
The islets of Langerhans are compact endocrine micro-organs scattered throughout the exocrine pancreatic parenchyma, comprising only 1–2% of the total pancreatic mass but receiving 10–15% of the gland's blood flow — reflecting their hormonal output priority.
Islet cell types and their products:
| Cell type | Proportion | Hormone | Primary action |
|---|---|---|---|
| β cells | ~65–80% | Insulin, C-peptide, amylin | Lowers blood glucose; anabolic (glycogen synthesis, lipogenesis, protein synthesis) |
| α cells | ~15–20% | Glucagon | Raises blood glucose (glycogenolysis, gluconeogenesis) |
| δ cells | ~5–10% | Somatostatin | Paracrine inhibitor of both insulin and glucagon |
| PP cells | ~1–2% | Pancreatic polypeptide | Inhibits exocrine secretion |
Insulin is synthesised as preproinsulin → cleaved to proinsulin → stored in secretory granules as insulin + C-peptide (connecting peptide). When β cells secrete insulin, C-peptide is released in equimolar amounts — making C-peptide a useful clinical marker of endogenous insulin secretion (exogenous insulin has no C-peptide).
Glucose-stimulated insulin secretion (GSIS): Glucose enters the β cell via GLUT2 → oxidative phosphorylation raises ATP:ADP ratio → ATP-sensitive K⁺ channel closes → membrane depolarisation → voltage-gated Ca²⁺ influx → exocytosis of insulin granules. This is the molecular target of sulfonylureas (which close the K⁺ channel pharmacologically).
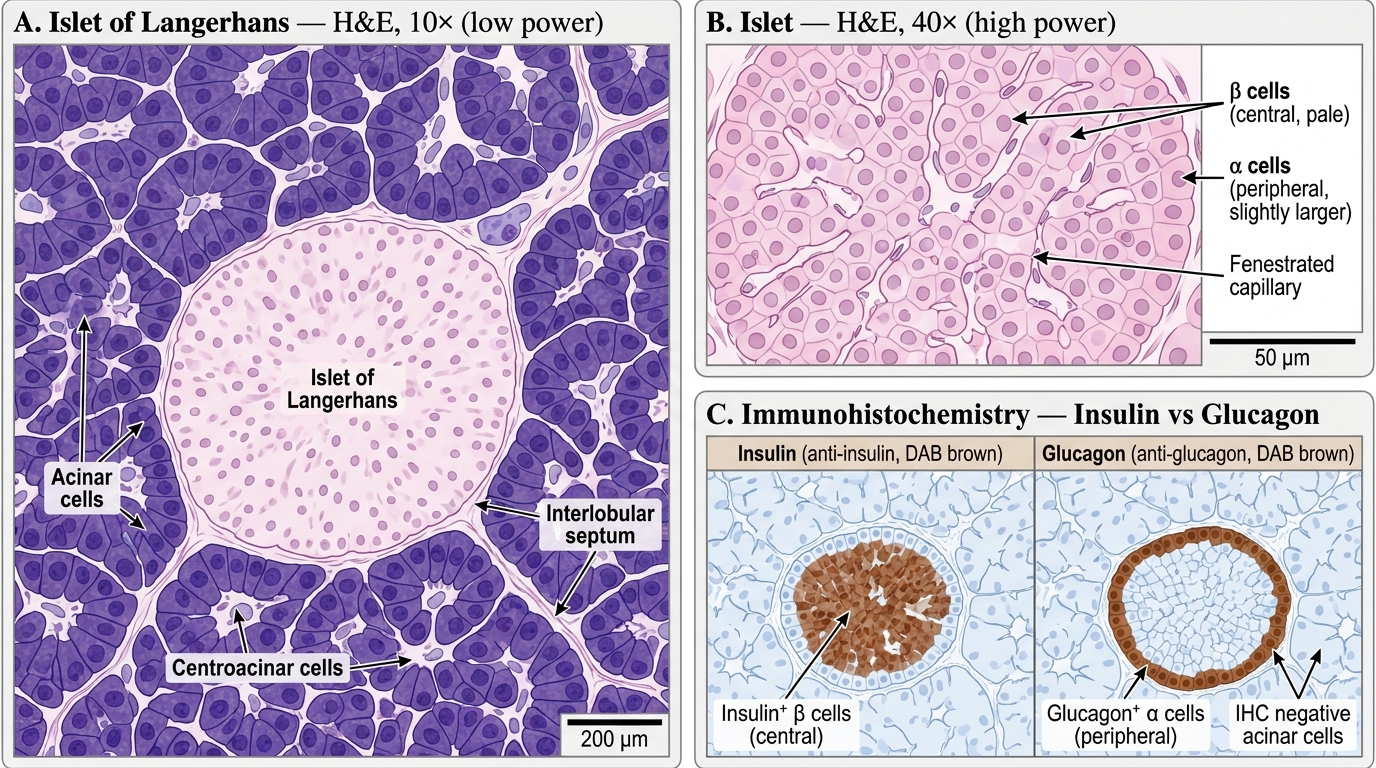
Islet of Langerhans — H&E and Immunohistochemistry
Classification of Diabetes Mellitus
Diabetes mellitus (DM) is a group of metabolic disorders characterised by chronic hyperglycaemia resulting from defects in insulin secretion, insulin action, or both, leading to impaired glucose homeostasis and progressive multisystem damage.
ADA/WHO Classification:
1. Type 1 DM (T1DM) — Immune-mediated β-cell destruction
- Accounts for ~5–10% of all DM
- Peak incidence: childhood/adolescence (but can present at any age)
- Absolute insulin deficiency → prone to diabetic ketoacidosis (DKA)
- Subtypes: 1A (autoimmune, most common) and 1B (idiopathic, non-autoimmune)
2. Type 2 DM (T2DM) — Insulin resistance + progressive β-cell failure
- Accounts for ~90–95% of all DM worldwide
- Older adults, but rising in younger age groups with obesity epidemic
- Relative insulin deficiency — DKA unusual; risk of HHS (hyperosmolar hyperglycaemic state)
3. Maturity-Onset Diabetes of the Young (MODY)
- Monogenic; autosomal dominant; strong family history; onset <25 years
- Six types (MODY 1–6), each caused by a single gene mutation affecting β-cell function
- Most common: MODY2 (glucokinase mutation — mild stable hyperglycaemia) and MODY3 (HNF1α — progressive, sulphonylurea-responsive)
- Often misclassified as T1DM or T2DM; correct diagnosis changes management
4. Secondary DM
- Pancreatic disease: chronic pancreatitis, cystic fibrosis, haemochromatosis (bronze diabetes), total pancreatectomy
- Endocrine: Cushing syndrome (↑cortisol), acromegaly (↑GH), phaeochromocytoma (↑catecholamines), glucagonoma
- Drug-induced: long-term corticosteroids, thiazide diuretics, anti-psychotics (atypicals → metabolic syndrome)
5. Gestational DM (GDM)
- Glucose intolerance first diagnosed in pregnancy (weeks 24–28 screening)
- Driven by placental hormones that antagonise insulin (hPL, progesterone, cortisol)
- Risk to foetus: macrosomia, neonatal hypoglycaemia, respiratory distress
- 50% of GDM mothers develop overt T2DM within 10 years
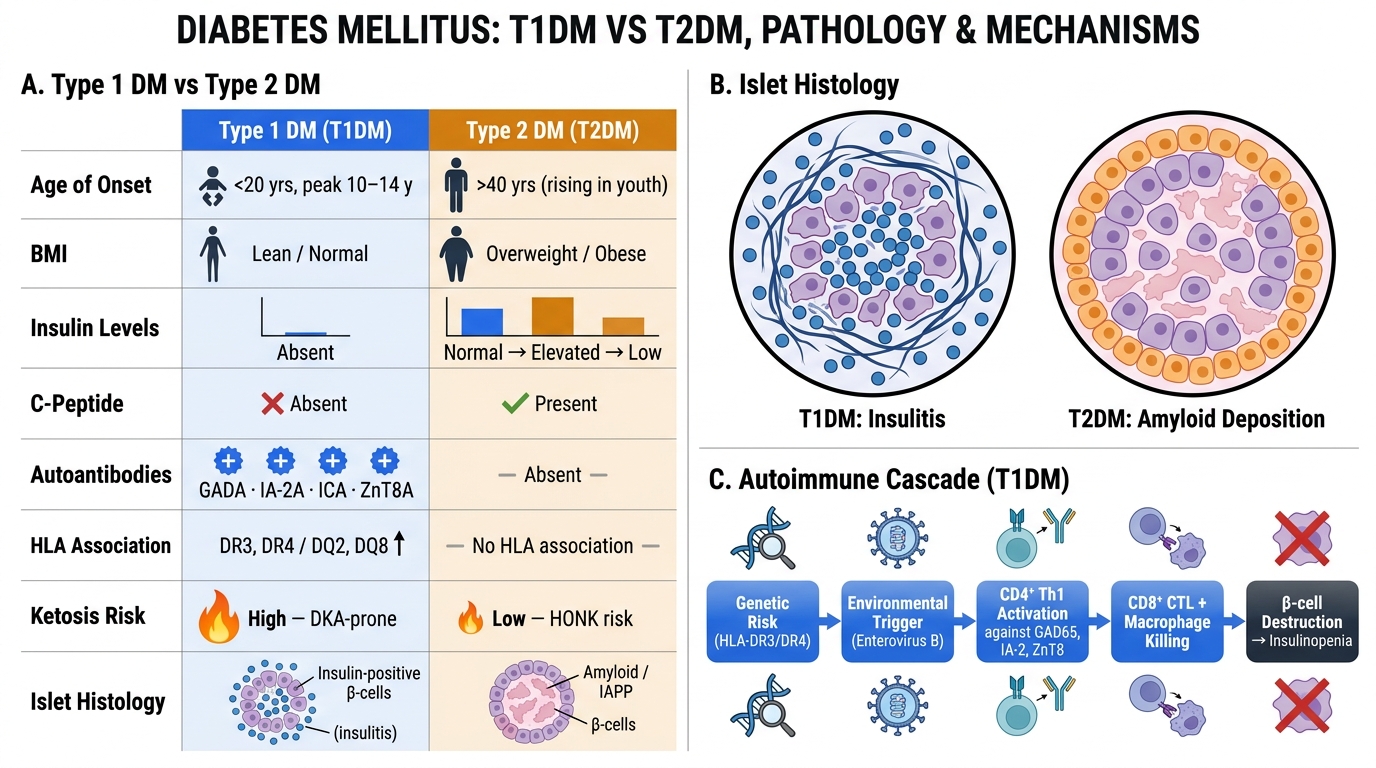
Type 1 DM vs Type 2 DM: Comparative Features, Islet Histology, and Autoimmune Pathogenesis
Type 1 DM — Autoimmune Pathogenesis
Type 1 DM is an organ-specific autoimmune disease in which the body's immune system selectively destroys the insulin-secreting β cells of the islets of Langerhans. The destruction is gradual but ultimately complete.
Genetic susceptibility — HLA association:
- ~95% of T1DM patients carry HLA-DR3, HLA-DR4, or both (haplotype HLA-DR3/DR4)
- HLA-DQ8 (linked to DR4) and HLA-DQ2 (linked to DR3) are the most specific risk alleles
- Protective allele: HLA-DQ6 (DQB10602) — its presence makes T1DM extremely unlikely
- The HLA class II molecules on antigen-presenting cells present (or fail to present/tolerise) islet peptides to CD4⁺ T helper cells — the starting point of the autoimmune cascade
- Non-HLA loci also contribute: INS gene (insulin promoter polymorphism), PTPN22 (lymphocyte phosphatase), CTLA4
Immunological cascade — the sequence of events:
1. An environmental trigger (viral infection — Enterovirus B is most implicated; also congenital rubella, dietary factors) in a genetically susceptible individual breaks central tolerance to islet antigens
2. CD4⁺ Th1 cells are sensitised against β-cell antigens (insulin, GAD65, IA-2, ZnT8)
3. CD4⁺ cells activate CD8⁺ cytotoxic T lymphocytes (CTLs) and macrophages → direct β-cell killing by perforin/granzyme and Fas-FasL pathways
4. Autoantibodies appear: anti-islet cell antibodies (ICA), anti-GAD65 (most sensitive), anti-insulin (IAA), anti-IA-2, anti-ZnT8 — these are markers* of the process, not the primary effectors
5. Gradual β-cell depletion (silent phase lasting months to years) until >80–90% of β cells are destroyed → clinical onset of DM
Insulitis — the histological hallmark:
- Islets show a dense lymphocytic infiltrate (predominantly T cells) during the active destruction phase
- Residual islets are small and fibrosed
- α, δ, and PP cells are spared
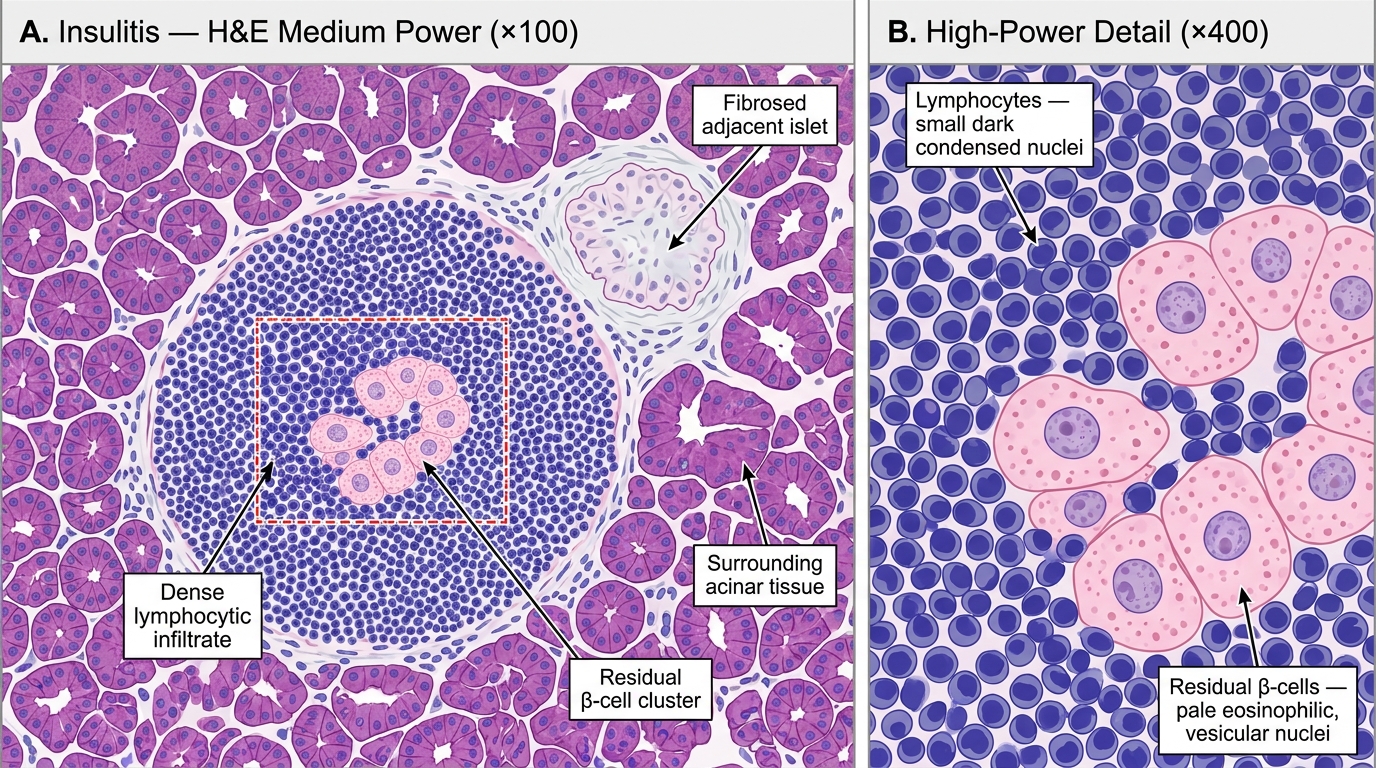
Insulitis in Type 1 Diabetes Mellitus — H&E Histology (Medium and High Power)
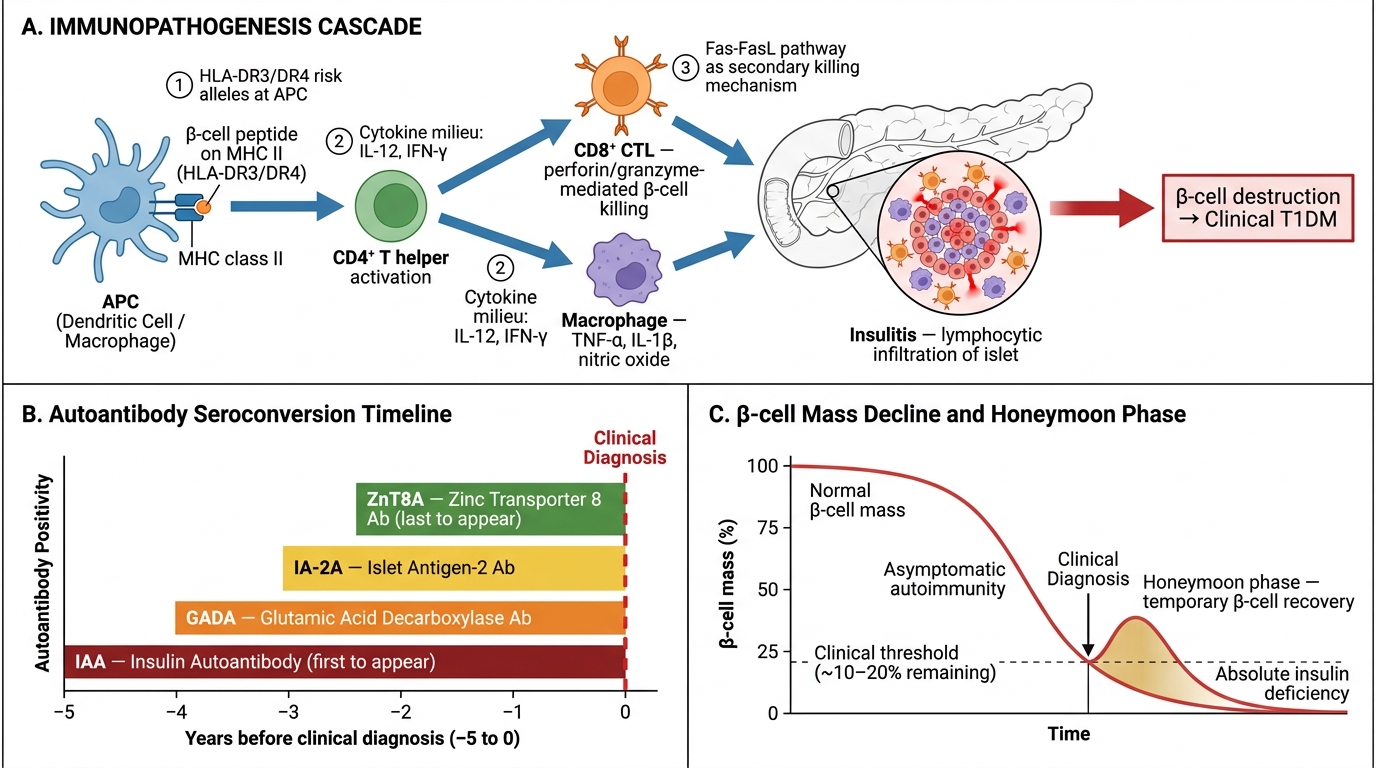
Immunopathogenesis of Type 1 Diabetes Mellitus — Cellular Cascade, Autoantibody Timeline, and β-cell Mass Kinetics
CLINICAL PEARL
The Honeymoon Phase of Type 1 DM: In the weeks after a T1DM diagnosis is treated with insulin, glucose control often improves dramatically and insulin requirements drop — sometimes to near-zero. This is the "honeymoon phase," lasting weeks to months. Residual β cells, relieved of glucotoxic stress, temporarily resume function. Parents and patients sometimes stop insulin, thinking the child is cured. They are not — the autoimmune destruction continues, and DKA recurs. The honeymoon phase is pathological confirmation that early insulitis is reversible up to a threshold, after which it is not.